Scientific Highlights
► A new quantum approach to solve electronic structures of complex materials► Quantum-Nuclear Effects in Amorphous Carbon
► Materials Simulation on Quantum Computer
► How to Transform Vacancies into Quantum Information
► Inorganic Nanocrystals Form Strongly-Coupled Supercrystals
► Machine Learning Accelerates Quantum Light-Matter Simulations
► Unraveling the Sectrets of Water through Quantum-mechanical Simulations
► Quantum Computing of Spin-Defect Properties
► Python Packages for First Principles Simulations
► Machine Learning Projects take Flight
► Two to Tango: Coupling Codes to Study Dissociation Chemistry
► Solar to Fuel: Interfacial Insights from First Principles
► Digging up Design Principles at the Buried Interfaces of Nanocrystalline Solids
► Unraveling the Thermodynamic Tug-of-War that forms Polyelectrolyte Complexes
► Curating and Exploring Scientific Papers with Qresp
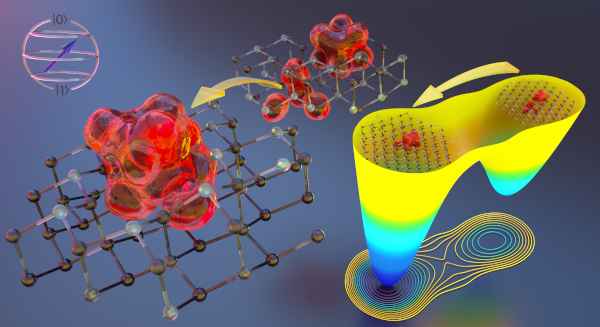
Unlike conventional computers that store information in binary bits, quantum computers use qubits that can exist in superposition of states, letting them solve certain problems more easily and quickly. Computational chemists are still debating whether and when quantum computers might eventually be able to tackle the electronic structure problem of complex materials better than conventional computers. However, today’s quantum computers remain relatively small and produce noisy data. Even with these weaknesses, MICCoM researchers wondered whether they still could make progress in creating the underlying quantum computational methods required to solve electronic structure problems on quantum computers. The researchers designed a hybrid simulation process, using IBM quantum computers. In their approach, a small number of qubits — between four and six — perform part of the calculations, and the results are then further processed using a classical computer. They were able to solve the electronic structure of realistic spin-qubits. Even if for now the electronic structures solved using the new quantum computational approach could already be solved using a conventional computer, the results obtained by the MICCoM team pave the way for quantum computers to address more complex chemical structures. The study was recently published in Journal of Chemical Theory and Computation and featured in Argonne National Lab News.



07/18/2022
In crystalline solids, atoms are arranged in periodic patterns on regular lattices. Amorphous solids, instead, lack long-range order; namely, a regular array of atoms beyond first or second nearest neighbors is absent. The lack of periodicity influences many properties of amorphous materials, including the coupling of electronic and nuclear motion. A recent study by MICCoM scientists studied amorphous carbon, a system composed of a relatively light atom. The authors showed that to understand its electronic properties, a quantum mechanical treatment of electron–nuclear coupling is essential, and illustrated a simulation framework based on first principles to do so. They also discussed the role of specific defect states in the disordered network in determining the physical properties of amorphous carbon. The work was recently published in PNAS and was featured in PME news.


05/24/2022
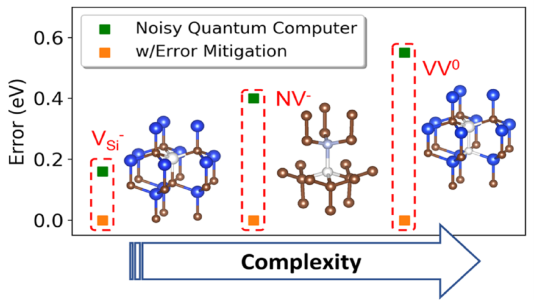
The jury is still out on whether quantum computers will be more efficient than classical ones in solving useful chemistry and physics problems. However, the promise held by storing information in quantum instead of classical bits is worth exploring, as quantum computers may lead, down the road, to revolutionary quantum simulations of solids and molecules. Recently, MICCoM researchers presented a computational protocol to study the electronic structure of spin qubits on a quantum computer. Spin qubits are point defects in semiconductors or insulators of interest for the realization of quantum technologies. The study presented a strategy describing a defect embedded in a periodic array of several hundred atoms representing a solid, using a relatively small number of degrees of freedom that are optimized on noisy quantum hardware. It then showed that hardware noise may substantially affect the results and ways are proposed to mitigate it and obtain energies of the defects that are in good agreement with those computed with a noiseless quantum simulator.The study was recently published in PRX Quantum and featured in Argonne National Lab News.
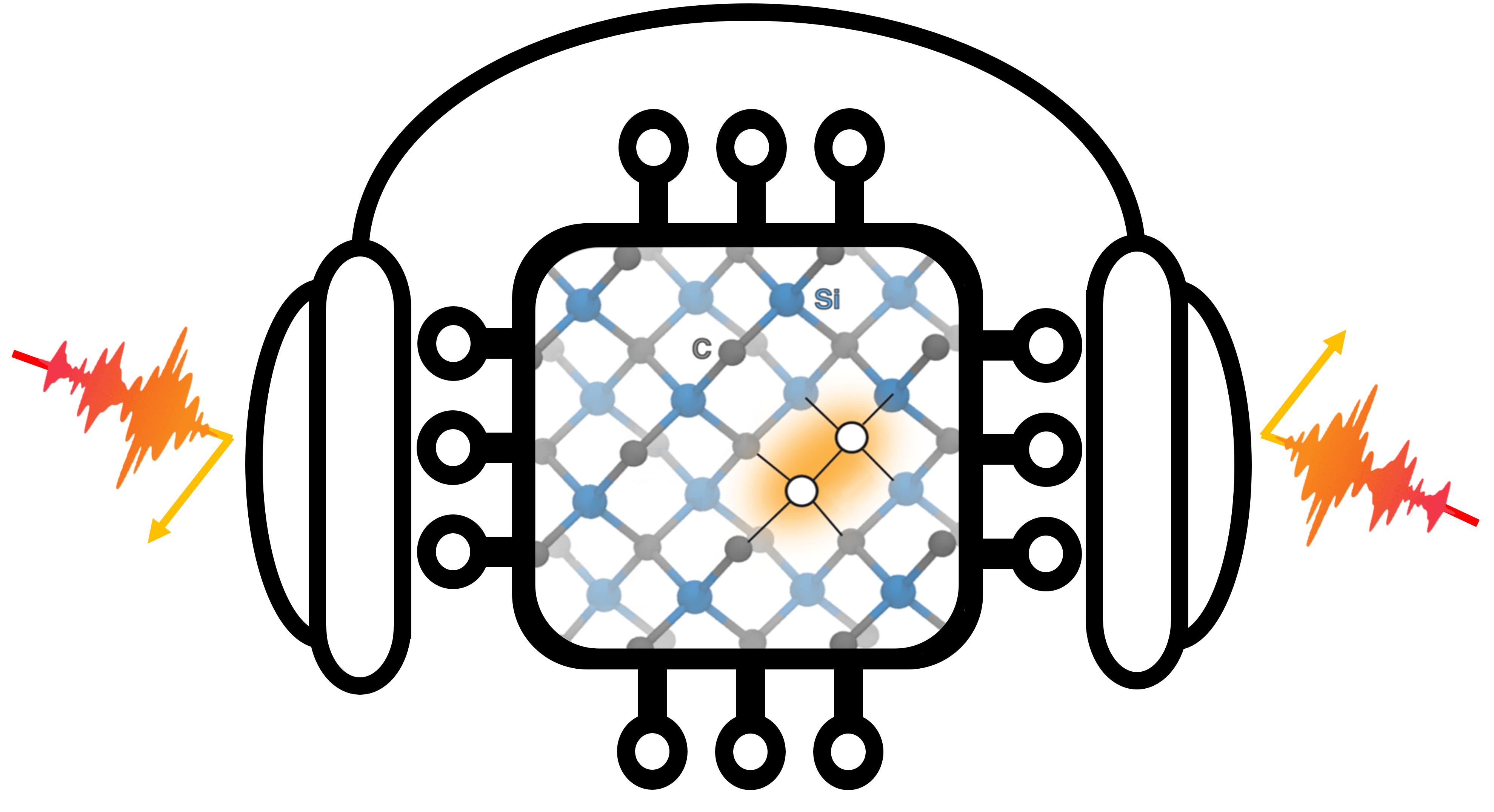
Divacancies in SiC are promising candidates for quantum-technology applications. However, little is known about how these divacancies form, move, and rotate. Using a combination of enhanced sampling, molecular dynamics, and density functional theory, MICCOM scientist recently revealed the formation mechanisms of divacancies in SiC. The authors not only determined that the formation of the divacancy is a thermally activated process competing with other defect conversion processes, but identified a different type of quasi-stable defect complex that had not been predicted before. The work, enabled by the coupling of various codes within MICCOM, was recently published in Nature Communications and featured in phys.org, Argonne News, and PME News



Growing collections of nanocrystals has
been performed previously with organic ligands. While this method
yields collections of nanocrystals, these are separated by
insulating barriers, which blocks the movement of electrons
between those crystals.
With a new method, recently presented in Science,
MICCOM scientists from the group of Dmitri Talapin uyse an all
inorganic ligand to produce ordered supercrystals with strong
electronic coupling. Their work requires a detailed understanding
of the structure and growth of these supercrystals on an atomic
scale, which is faciliated by combining powerful x-ray
spectroscopy with intricate simulation and modeling of the
underlying physics and chemistry.
By controling the chemical conditions during growth, the authors
showed a fine-tuned control of the electronic properties of the
supercrystals and demonstrated fully reversible growth.



Quantum simulations of light-matter interactions are important
tools to predict optimal materials for photovoltaics and
photelectrochemical cells. However, these simulations are
demanding from a computational standpoint, especially when applied
to complex, heterogeneous systems. Recently
MICCOM and AMEWS researchers
developed a novel formalism to accelerate these simulations
using machine-learning algorithms, Their work was recently
published in Chemical
Science and featured in
and UChicago PME News.

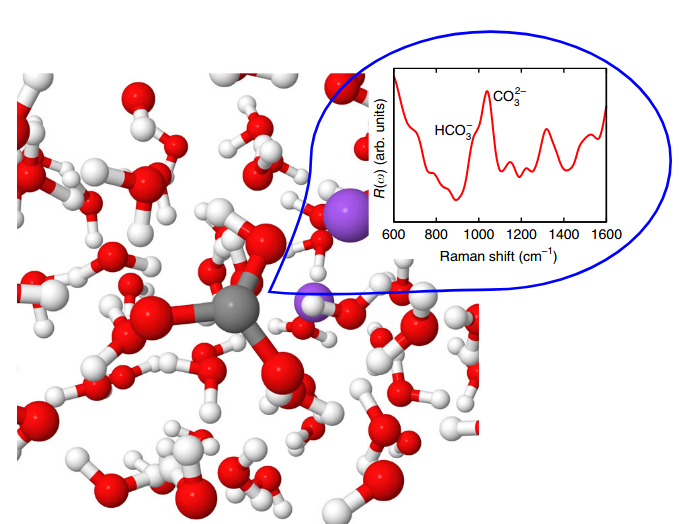
Water is ubiquitous on earth, and the knowledge how matter
interacts with water is paramount for a wide range of
applications, from climate science to energy engineering.
Recently, MICCoM scientists studied two very different
interactions of water with matter: Ding Pang and Giulia Galli
determined the properties of dissolved carbon in water under
high pressure in the earth's mantle from first-principles
simulations. Understanding what concentration of carbon can be
found under these extreme conditions has important implications
on how carbon moves from the atmosphere to the earth's mantle.
Their
study, recently featured in UChicago
PME news, found that the carbon concentration deep in the
mantle was underestimated by previous models.
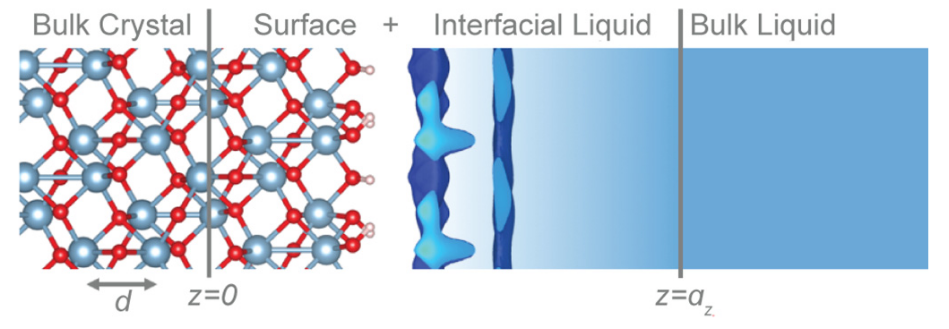 In another
study of water-matter interactions, MICCoM scientists
partnered with scientists at the Argonne National Laboratory to
validate first-principles calculations of water/metal-oxide
interfaces. These play a crucial role in a wide range of
applications, such as photochemical water splitting and
electrochemical carbon-dioxide reduction. In a combined
theoretical and experimental study, the scientists established a
groundbreaking protocol to validate the first-principles
simulations. This research provides a pathway for more accurate
models of water/metal-oxide interfaces in the future.
In another
study of water-matter interactions, MICCoM scientists
partnered with scientists at the Argonne National Laboratory to
validate first-principles calculations of water/metal-oxide
interfaces. These play a crucial role in a wide range of
applications, such as photochemical water splitting and
electrochemical carbon-dioxide reduction. In a combined
theoretical and experimental study, the scientists established a
groundbreaking protocol to validate the first-principles
simulations. This research provides a pathway for more accurate
models of water/metal-oxide interfaces in the future.


03/19/2021
Quantum computing promises to greatly advance our capability to
calculate properties of molecules and solids beyond the limits
of current supercomputers. However, quantum computing is still
in it's infancy and the necessary hardware and algorithms are
still being developed. MICCoM scientists have recently developed
a new quantum-mechanical
framework to use quantum computing to determine the
properties of spin defects. Combining calculations on classical
and quantum hardware, they demonstrated that even the currently
available imperfect quantum computers yield accurate and effective
results for these complex systems. The new framework, developed by
He Ma, Nan Sheng, Marco Govoni, and Giulia Galli, was recently
published in a series of papers in npj
Computational Materials, Journal of
Physical Chemistry and Chemical Physics, and Journal
of Chemical Theory and
Computation, and was
featured in a Argonne
National Laboratory press release.



MICCoM has released two python packages for analysis of first principles simulations: PyCDFT (code repository), and PyZFS (code repository) . PyCDFT computes diabatic states using constrained density functional theory (CDFT). It provides an object-oriented, customizable implementation of CDFT, and allows for both single-point self-consistent-field calculations and geometry optimizations. PyZFS, on the other hand, is used for the calculation of the zero-field splitting tensor, D, of molecules and solids, based on wavefunctions obtained from density functional theory calculations. PyZFS features a modular design and utilizes abstract classes for extensibility. It has already been used to predit ZFS tensors for spin defects in semiconductors as well as spin-phonon interactions in solids. Both PyCDFT and PyZFS can use wavefunctions generated by various plane-wave DFT codes as input, including

12/19/2019
As machine learning (ML) techniques become increasingly ascendant in
computational materials science, MICCoM is developing expertise and successfully
launched several projects in this field. MICCoM scientists from the group of
Prof. Juan de Pablo published a Science Advances paper on using supervised machine learning to
obtain electronic structure at coarse-grained resolution. The work was featured by
Argonne and UChicago News. It uses artificial neural networks and
coarse-graining to compute electronic structure across various molecular
conformations. The method was later applied to study optoelectronic properties
of conjugated polymers, published in Macromolecules. 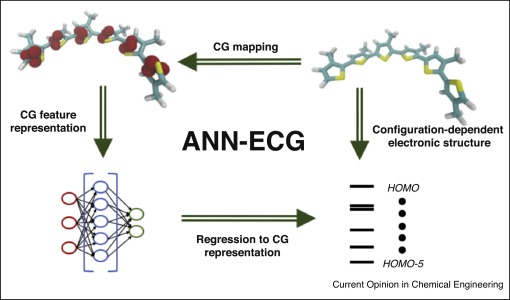 MICCoM researchers have also contributed perspective
articles towards the application of machine learning to collective variables
in biomolecular simulation, published in
Molecular Physics, as well
as multiscale soft materials design,
published in
Current Opinion in Chemical
Engineering. In addition,
MICCoM PI Andrew Ferguson was also
recently featured in a UChicago news
story on "How AI could change
science," where he described how
using physical laws as inputs to
artificial intelligence algorithms
could accelerate discovery of new
materials. Finally, ML techniques are
being applied to electronic structure
problems as well, for example the calculation of
absorption spectra.
MICCoM researchers have also contributed perspective
articles towards the application of machine learning to collective variables
in biomolecular simulation, published in
Molecular Physics, as well
as multiscale soft materials design,
published in
Current Opinion in Chemical
Engineering. In addition,
MICCoM PI Andrew Ferguson was also
recently featured in a UChicago news
story on "How AI could change
science," where he described how
using physical laws as inputs to
artificial intelligence algorithms
could accelerate discovery of new
materials. Finally, ML techniques are
being applied to electronic structure
problems as well, for example the calculation of
absorption spectra.




The investigation of salts in water at extreme conditions is crucial to
understanding the properties of aqueous fluids in the Earth. In this paper, published in Nature
Communications, MICCoM scientists report first principles and
classical molecular dynamics simulations of NaCl at temperatures and pressures
relevant to the Earth’s upper mantle. The work was UChicago PME news. The study employed the codes



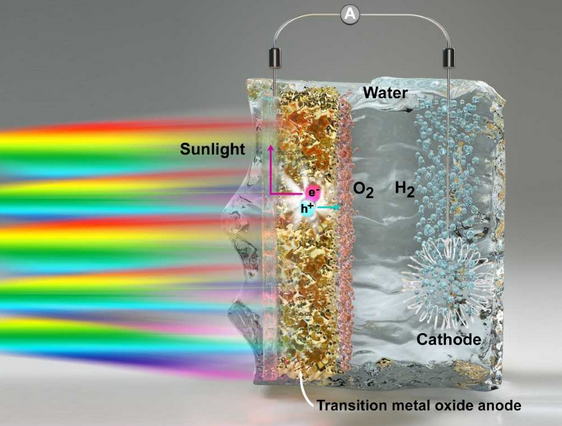
The design and optimization of photoelectrodes for artificial photosynthesis are critical to achieving sustainable solar to fuel conversion technologies. Most computational screening of materials so far has focused on bulk properties, leaving the critical water/photoanode interface poorly understood. In this paper, published in Nature Materials, MICCoM scientists employed first-principles codes developed in the center to investigate a promising photoanode for water oxidation, tungsten trioxide (WO3). They identified three major factors determining the chemical reactivity of the material interfaced with water: the presence of surface defects, the dynamics of excess charge at the surface, and finite temperature fluctuations of the surface electronic orbitals. Being generalizable properties, these key computational insights will aid the interfacial molecular engineering of oxide photoabsorbers for water oxidation.
This study employed the MICCoM codes
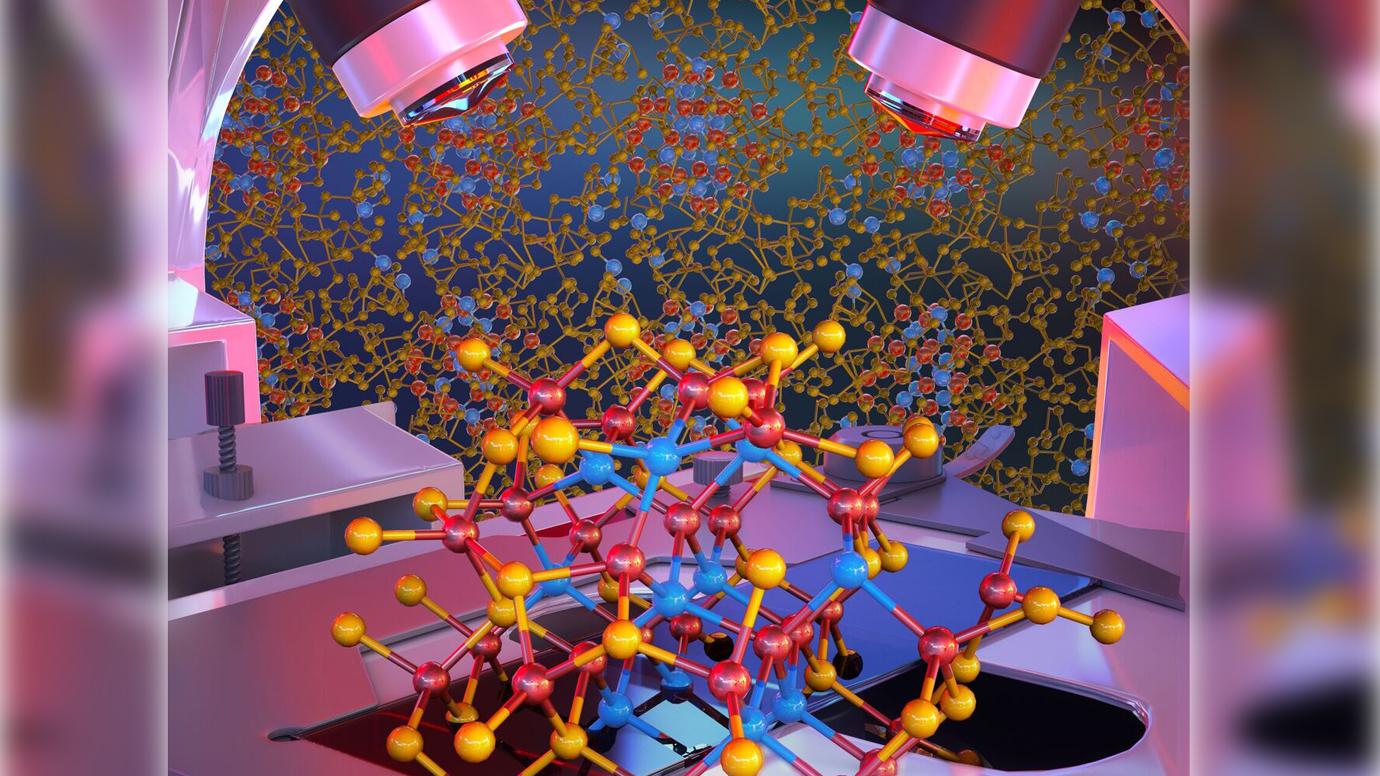
Semiconducting nanomaterials are critical to emerging optoelectronic and photonic technologies. In such systems, understanding surface chemistry of the interface between nano building blocks and their ligands is an outstanding challenge for rational device engineering. In this study, published in Nature Nanotechnology, a joint computational/experimental team of MICCoM scientists presented an approach to characterize such buried interfaces in solids of prototypical InAs nanoparticles capped with Sn2S64– ligands. They discovered that that inorganic ligands dissociate on InAs to form a surface passivation layer. A nanocomposite with unique electronic and transport properties is formed, exhibiting type II heterojunctions favourable for exciton dissociation. With these insights, they establish crucial design principles towards attaining desirable electronic and transport properties, while also explaining the origin of unusual measured negative photoconductivity of the nanocrystalline solids.
This study employed the MICCoM code


Polyelectrolytes are "smart" materials that find a variety of applications including drug delivery, gene therapy, layer-by-layer films, and fabrication of ion filtration membranes. They fall into two distinct categories - strong or weak - based on their complete or partial ionization in solution, forming polyelectrolyte complexes. However, the thermodynamic origins of polyelectrolyte complex formation have been poorly understood.
In this study, published in Journal of the American Chemical Society, MICCoM scientists employed coarse-grained molecular dynamics simulations to study the thermodynamics at play in the complexation processes. They used an expanded-ensemble method to explicitly separate entropic and enthalpies contributions to free energy. In so doing, they discovered that strong polyelectrolytes are primarily driven by entropic effects, consistent with experimental studies. The effect was attributed to gain in entropy from the release of counterions upon complex formation. On the other hand, they found that complex formation for weak polyelectrolytes, where counterions are not as strongly bound, is more influenced by enthalpic effects. Their findings will guide the molecular engineering of tunable, weak polyelectrolytes for various applications in molecular capture and release and layer-by-layer assembly of films.
This study employed the MICCoM code




The burgeoning age of high performance computing and big data is revolutionizing both the methods and pace of scientific research. However, the development of digital data infrastructures has not kept up with this rapid growth, especially in the field of computational research for energy application. The community sorely needs open-source strategies for documenting codes, curating workflows, and publishing computational data. In view of these challenges, MICCoM has created the open source software Qresp: “Curation and Exploration of Reproducible Scientific Papers." In this study, published in Scientific Data, MICCoM scientists introduce Qresp in detail. The tool allows users to curate the codes/data of a completed publication, while storing all data locally. Towards this end, Qresp has an intuitive graphical user interface (GUI) designed to simply prompt user input information about a publication in order to generate metadata in JSON format. On the other hand, once a paper and its codes/data are curated, Qresp's GUI may also be used as a searchable explorer of metadata or a means to dive into the entire workflow and procedures of an individual paper, from code inputs to finished figures. The Qresp project is one of the cornerstones of the MICCoM data effort. Many publications from the center have already been curated with Qresp and are being used for both validation efforts and pedagogy.
