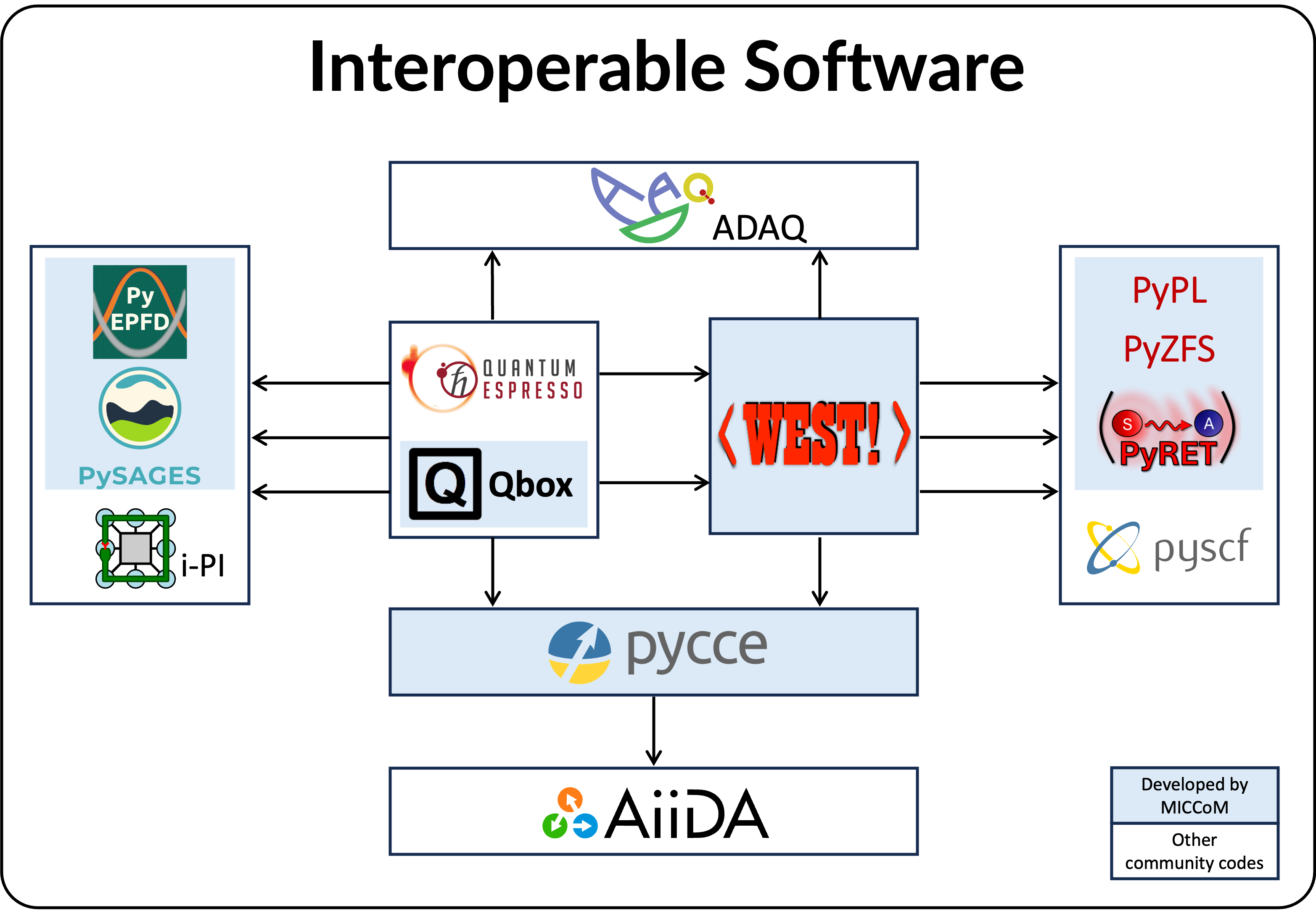
Software
Qbox:
First-principles molecular dynamics
Code repository
WEST:
Many-body perturbation theory
Code repository

PySAGES:
Reactive pathway identification and free energy
calculation
Code repository; SSAGES repository

PyCCE:
Dynamics of spin qubits interacting with a spin bath
Code repository

PyRET:
resonance energy transfer processes between localized defects in solids
Code repository
-
Qbox is a first principles molecular dynamics (MD) code using Density Functional Theory (DFT) and hybrid-DFT. In addition to the simulation of thermodynamic and structural properties of materials, Qbox allows for the calculations of vibrational spectra, ionic conductivity and heat transport coefficients.
- WEST performs large-scale many-body perturbation theory calculations providing electronic and optical spectroscopic characterization of complex materials.
- PySAGES is a GPU-enabled version of SSAGES. PySAGES and SSAGES provide a comprehensive suite of advanced methods for phase space sampling organized into a unified framework that can be used as a wrapper for quantum and classical MD and Monte Carlo (MC) engines. They are coupled with several MD and electronic structure codes.
- PyCCE (code repository) is an open-source python library to simulate the dynamics of spin qubits interacting with a spin bath using the cluster-correlation expansion (CCE) method.
- PyZFS (code repository), PyEPFD (code repository), PyPL (code repository) , and PyRET (code repository) are python packages coupled to first principles simulations and electronic structure calculation engines. PyZFS is used for the calculation of the zero-field splitting (ZFS) tensor, D, of molecules and solids, based on wavefunctions obtained from density functional theory calculations. PyEPFD is a Python library to compute electron-phonon interactions from finite displacements (using either frozen phonon and or stochastic methods) and to analyze molecular dynamics trajectories and determine electron-phonon interactions. PyPL computes vibrationally resolved optical spectra of point defects using atomic positions and phonons obtained from DFT calculations. PyRET enables calculations of non-resonant energy transfer processes.
- WEST and Qbox, PySAGES / SSAGES and Qbox, and Qbox and i-PI are coupled in client-server mode. For details, see npj Comput. Mater. 7, 32, (2021). DOI: 10.1038/s41524-021-00501-z.
- Qresp: "Curation and Exploration of Reproducible Scientific Papers" is a graphical user interface (GUI) that allows to curate and explore data presented in scientific publications, including the creation of workflows.
- WEST performs large-scale many-body perturbation theory calculations providing electronic and optical spectroscopic characterization of complex materials.