Publications
Analytical Forces from the Bethe-Salpeter Equation for Large-Scale Excited-State Relaxation
Yu Jin, Victor Wen-zhe Yu, Marco Govoni, Giulia Galli
Submitted (2026) DOI: 10.48550/arXiv.2607.20728
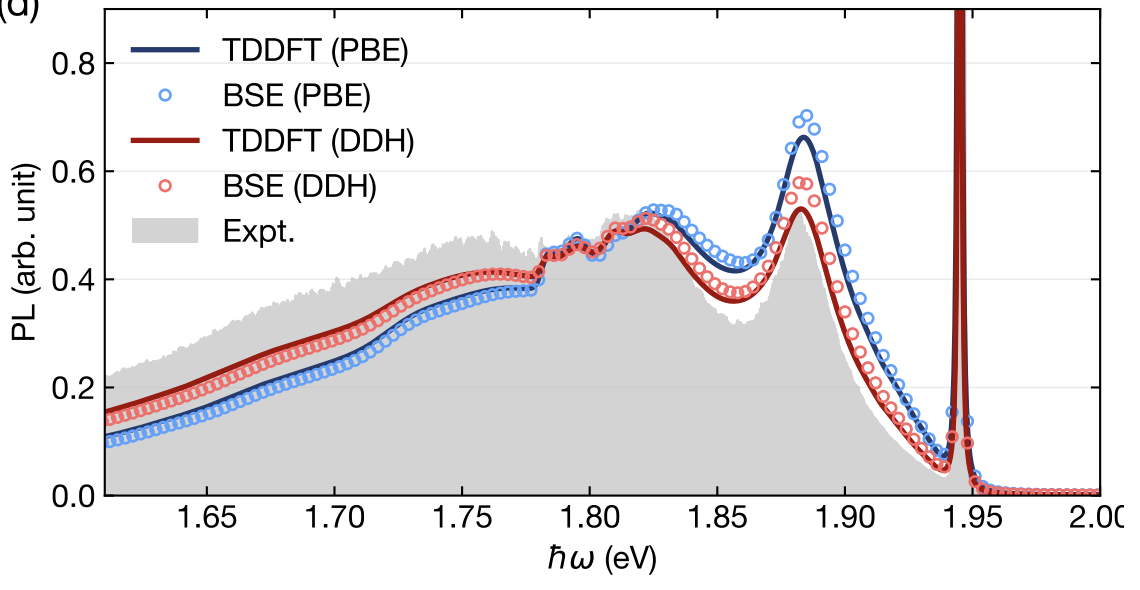
We present an efficient plane-wave implementation of analytical nuclear forces for electronic excited states described by the Bethe-Salpeter equation (BSE). The formulation combines density-matrix perturbation theory with a Lagrangian approach, and avoids both explicit empty-state summations and the response calculations for each atomic displacement, required by conventional approaches based on density functional perturbation theory. Together with GPU acceleration, these advances make BSE forces calculations tractable for solid-state systems containing hundreds of atoms. We demonstrate the method on two point defects with distinct dielectric environments: the nitrogen-vacancy center in diamond, where BSE and time-dependent density functional theory (TDDFT) yield consistent excited-state relaxations, and the carbon-dimer defect in two-dimensional hexagonal boron nitride, where the screened electron-hole interaction included in the BSE stabilizes the localized defect excitation and corrects the relaxation pattern predicted by semilocal TDDFT. These results establish a scalable framework for BSE-level studies of excited-state relaxation and vibronic coupling in heterogeneous condensed systems.

The WEST code for large-scale excited-state materials simulations
Victor Wen-zhe Yu, Siyuan Chen, Yu Jin, Vrindaa Somjit, Stefano Paolo Villani, Jiawei Zhan, Marco Govoni, Giulia Galli
Submitted (2026) DOI: 10.48550/arXiv.2607.14025
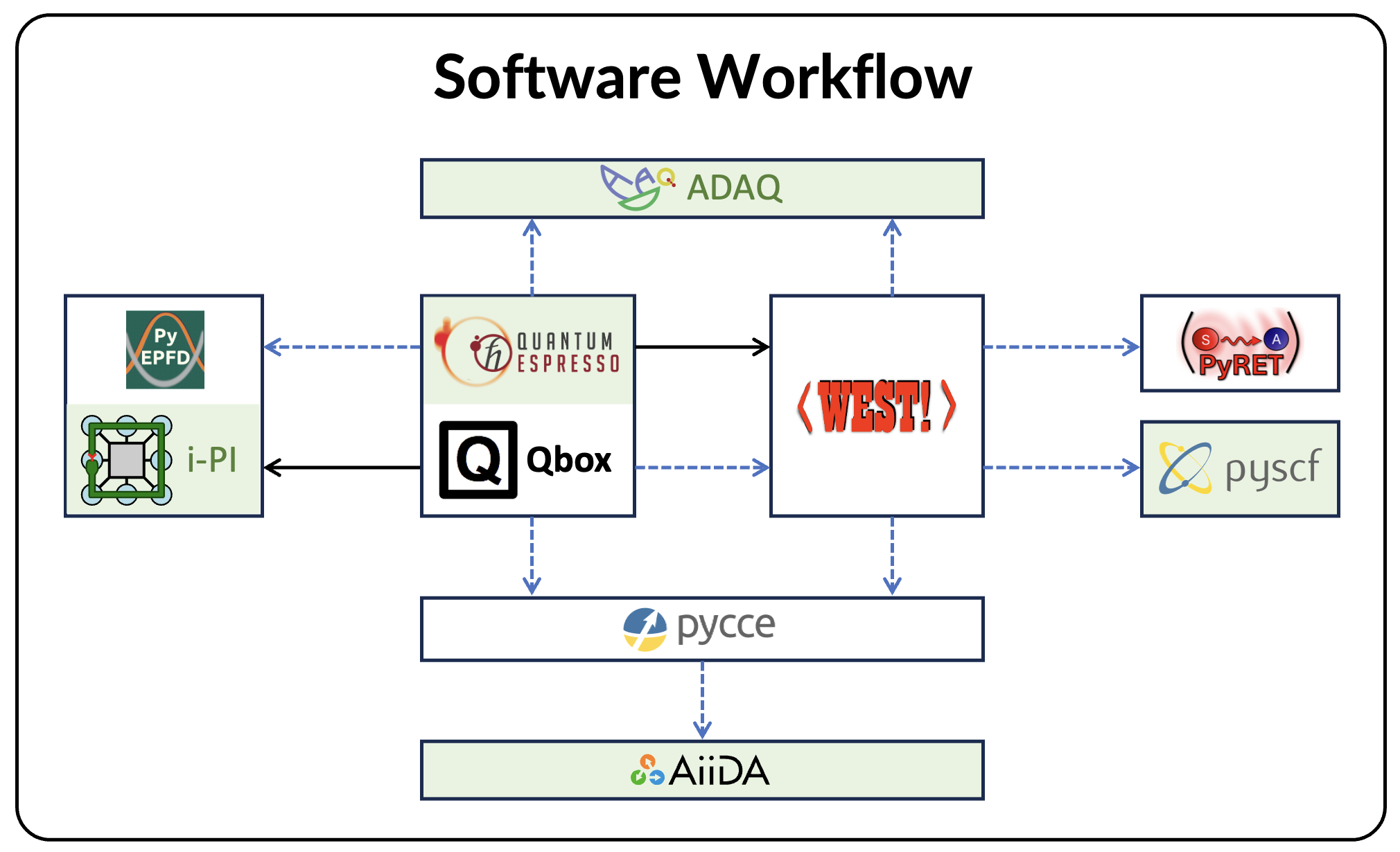
We present WEST, an open-source plane-wave pseudopotential code for large-scale excited-state materials simulations, and describe its theoretical foundations, software architecture, and capabilities. WEST implements full-frequency GW, quantum defect embedding theory, the Bethe-Salpeter equation, and time-dependent density functional theory within a common algorithmic framework that avoids the explicit computation of virtual electronic states. By combining density functional and density matrix perturbation theory, low-rank representations of the dielectric screening and exact exchange, and localization techniques, WEST achieves favorable computational scaling with system size. The code supports the calculation of quasi-particle and neutral excitation energies, optical and photoluminescence spectra, excited-state forces, and non-adiabatic couplings, with interoperable workflows connecting to quantum chemistry, vibronic coupling, and quantum computing packages. A hierarchical parallelization strategy and GPU acceleration deliver near-ideal strong scaling to thousands of GPUs, enabling accurate excited-state simulations of systems with more than a thousand atoms. Representative applications, spanning the full optical cycle of solid-state spin defects, self-trapped excitons in metal-halide perovskites, and the optical response of liquid water and ice, demonstrate the accuracy and versatility of the code across diverse material classes. The capabilities implemented in WEST establish the code as a scalable platform for predictive excited-state simulations, high-throughput materials discovery, and the generation of high-fidelity datasets for machine learning in computational materials science.
First-principles calculations of internal conversion processes in spin defects
Stefano Paolo Villani, Yu Jin, Giulia Galli
Submitted (2026) DOI: 10.48550/arXiv.2606.14545
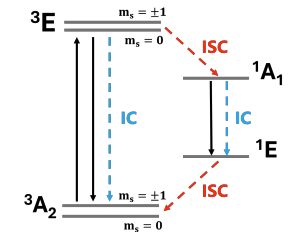
Optically active spin defects are foundational for quantum technologies, yet common approximations underestimate their internal conversion (IC) rates by orders of magnitude. We propose a broad, predictive framework to compute IC rates that incorporates multi-configurational effects via many-body wavefunctions in TDDFT, and includes all-phonon-mode contributions via analytical non-adiabatic couplings. Our approach resolves discrepancies with experiment, achieving quantitative agreement for the NV− center in diamond, and identifying a previously overlooked non-radiative channel in the divacancy triplet lifetime in SiC.
Spin Dynamics from Atomistic Quantum Simulations
Enrico Drigo, Marquis M. McMillan, Benjamin Pingault, Yinan Dong, F. Joseph Heremans, David D. Awschalom, Giulia Galli
Submitted (2026) DOI: 10.48550/arXiv.2605.04294
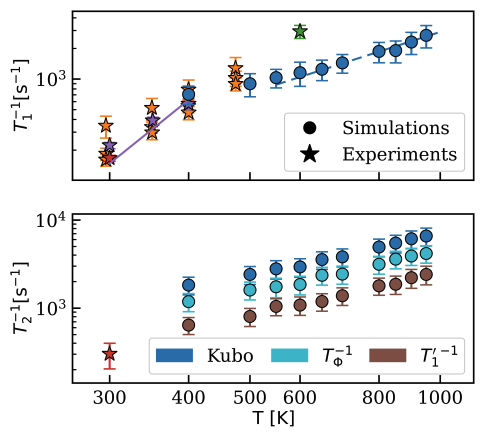
Optically active solid-state spin defects are promising candidates for quantum applications, however a unified theoretical framework to predict their spin dynamics at high temperatures is not yet available. Here, using Kubo linear-response theory, we derive expressions of spin-lattice and decoherence times T_1 and T_2 in terms of correlation functions of spin-lattice couplings. We then evaluate T_1 and T_2 from molecular dynamics and spin-lattice interaction time-series generated by state-of-the-art machine learning models trained on ab initio data. Finally we measure T_1 times for the NV center in diamond and compare experimental and theoretical results, showing excellent agreement.
Strategies to predict and design spin defects for quantum technologies
Giulia Galli, Alfonso Castillo, Swarnabha Chattaraj, Siyuan Chen, Marco Govoni, Yu Jin, Jonah Nagura, Abigail N Poteshman, Vrindaa Somjit, Michael Y Toriyama, Victor Wen-zhe Yu, and Cunzhi Zhang
J. Chem. Theory Comp. Accepted (2026), Editors' Choice. DOI: 10.1021/acs.jctc.6c00670
The design of materials with functionalities optimally suited for quantum information applications is a critical need in the field of quantum science and engineering. This Perspective focuses on a specific class of systems, spin defects in semiconductors and insulators, and on the manipulation of their electron spins, which can provide controllable qubits with long relaxation and coherence times, and can be coupled to nuclear spins for long-lived quantum memories. We summarize our recent contributions to the development of integrated theoretical frameworks and high-performance codes to predict and design spin defects and present examples of validated predictions and interpretations of experimental results. Starting from a brief description of the structural and charge stability at zero temperature using density functional theory, we present simulations to understand the mechanism of spin defect formation with first-principles molecular dynamics and machine learned potentials. We then discuss two classes of properties that are essential for the prediction of spin defects' functionalities: electronic and coherence properties. The discussion of computational frameworks is followed by that of results for specific systems illustrating successes, open problems, and future applications, with examples for heterogeneous solids, inclusive of surfaces and mesoscopic defects, and with a focus on quantum sensing and communication applications.
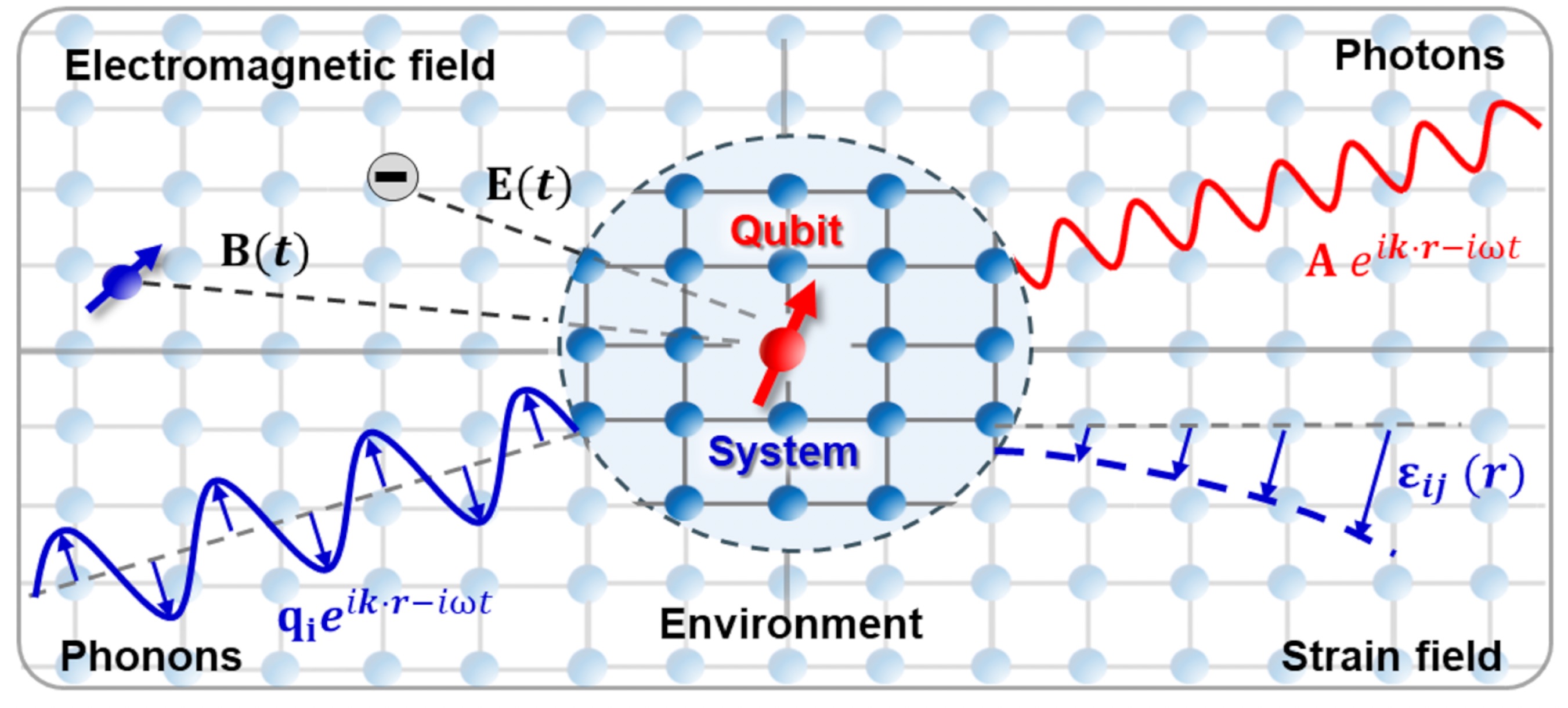
Optimizing spin qubit coherence through materials codesign
Vrindaa Somjit*, Gregory Grant*, Swarnabha Chattaraj*, Supratik Guha, David D. Awschalom, Giulia Galli, Jiefei Zhang, and F. Joseph Heremans (*equal contribution)
MRS Bulletin 51, 260-275 (2026) DOI: 10.1557/s43577-026-01069-z
The evolution of defect-based spin qubit systems is currently transitioning from fundamentalstudies and proof-of-concept demonstrations into applications in the burgeoning field ofquantum technology. Within this context, new challenges emerge, in particular, the needto understand and engineer the fundamental materials that form the hardware buildingblocks critical for the scalability and wide-scale adoption of such technologies. While earlierdiscussions have often focused on qubits within idealized systems, major limitations onspin coherence and optical properties arise from effects imposed by the nonideality of thesurrounding host matrix. Decoherence can stem from a variety of sources, including otherqubits, nuclear spins, and parasitic point- and extended defects, which interact with the qubitvia magnetic and electric fields, photons, phonons, and strain. In this article, we focus on therelevant sources and mechanisms through which decoherence occurs and provide potentialmitigation strategies via the synergistic integration of first-principles simulations and materialssynthesis and engineering. We aim to provide a tangible link between material properties andmaterial functions thereby enabling materials-by-design.
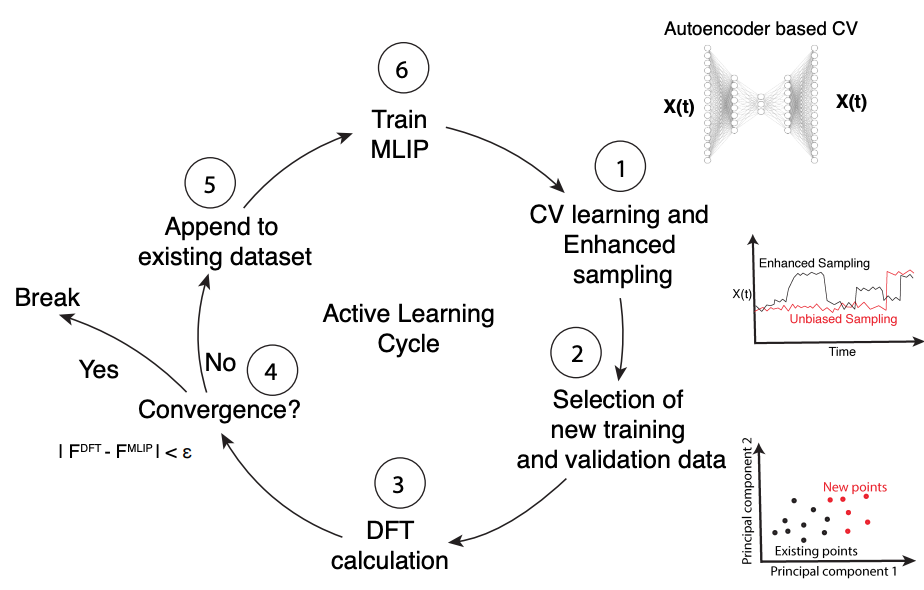
Characterizing Defect Dynamics in Silicon Carbide Using Symmetry-Adapted Collective Variables and Machine Learning Interatomic Potentials
Soumajit Dutta, Cunzhi Zhang, Gustavo Perez Lemus, Juan J. de Pablo, Francois Gygi, Giulia Galli, Andrew L. Ferguson
J. Chem. Theory Comp. 22, 9, 4728-4741 (2026) DOI: 10.1021/acs.jctc.6c00063
Silicon carbide (SiC) divacancies are attractive candidates for spin defect qubits possessing long coherence times and optical addressability. The high activation barriers associated with SiC defect formation and motion pose challenges for their study by first-principles molecular dynamics. In this work, we develop and deploy machine learning interatomic potentials (MLIPs) to accelerate defect dynamics simulations while retaining ab initio accuracy. We employ an active learning strategy comprising symmetry-adapted collective variable discovery and enhanced sampling to compile configurationally diverse training data, calculation of energies and forces using density functional theory (DFT), and training of an E(3)-equivariant MLIP based on the Allegro model. The trained MLIP reproduces DFT-level accuracy in defect transition activation free energy barriers, enables the efficient and stable simulation of multi-defect 216-atom supercells, and permits an analysis of the temperature dependence of defect thermodynamic stability and formation/annihilation kinetics to propose an optimal annealing temperature to maximally stabilize VV divacancies.
Trans-dimensional Hamiltonian model selection and parameter estimation from sparse, noisy data
Abigail N. Poteshman, Jiwon Yun, Tim H. Taminiau, Giulia Galli
Quantum 10, 2055 (2026). DOI: 10.22331/q-2026-04-08-2055
High-throughput characterization often requires estimating parameters and model dimension from experimental data of limited quantity and quality. Such data may result in an ill-posed inverse problem, where multiple sets of parameters and model dimensions are consistent with available data. This ill-posed regime may render traditional machine learning and deterministic methods unreliable or intractable, particularly in high-dimensional, nonlinear, and mixed continuous and discrete parameter spaces. To address these challenges, we present a Bayesian framework that hybridizes several Markov chain Monte Carlo (MCMC) sampling techniques to estimate both parameters and model dimension from sparse, noisy data. By integrating sampling for mixed continuous and discrete parameter spaces, reversible-jump MCMC to estimate model dimension, and parallel tempering to accelerate exploration of complex posteriors, our approach enables principled parameter estimation and model selection in data-limited regimes. We apply our framework to a specific ill-posed problem in quantum information science: recovering the locations and hyperfine couplings of nuclear spins surrounding a spin-defect in a semiconductor from sparse, noisy coherence data. We show that a hybridized MCMC method can recover meaningful posterior distributions over physical parameters using an order of magnitude less data than existing approaches, and we validate our results on experimental measurements. More generally, our work provides a flexible, extensible strategy for solving a broad class of ill-posed inverse problems under realistic experimental constraints.
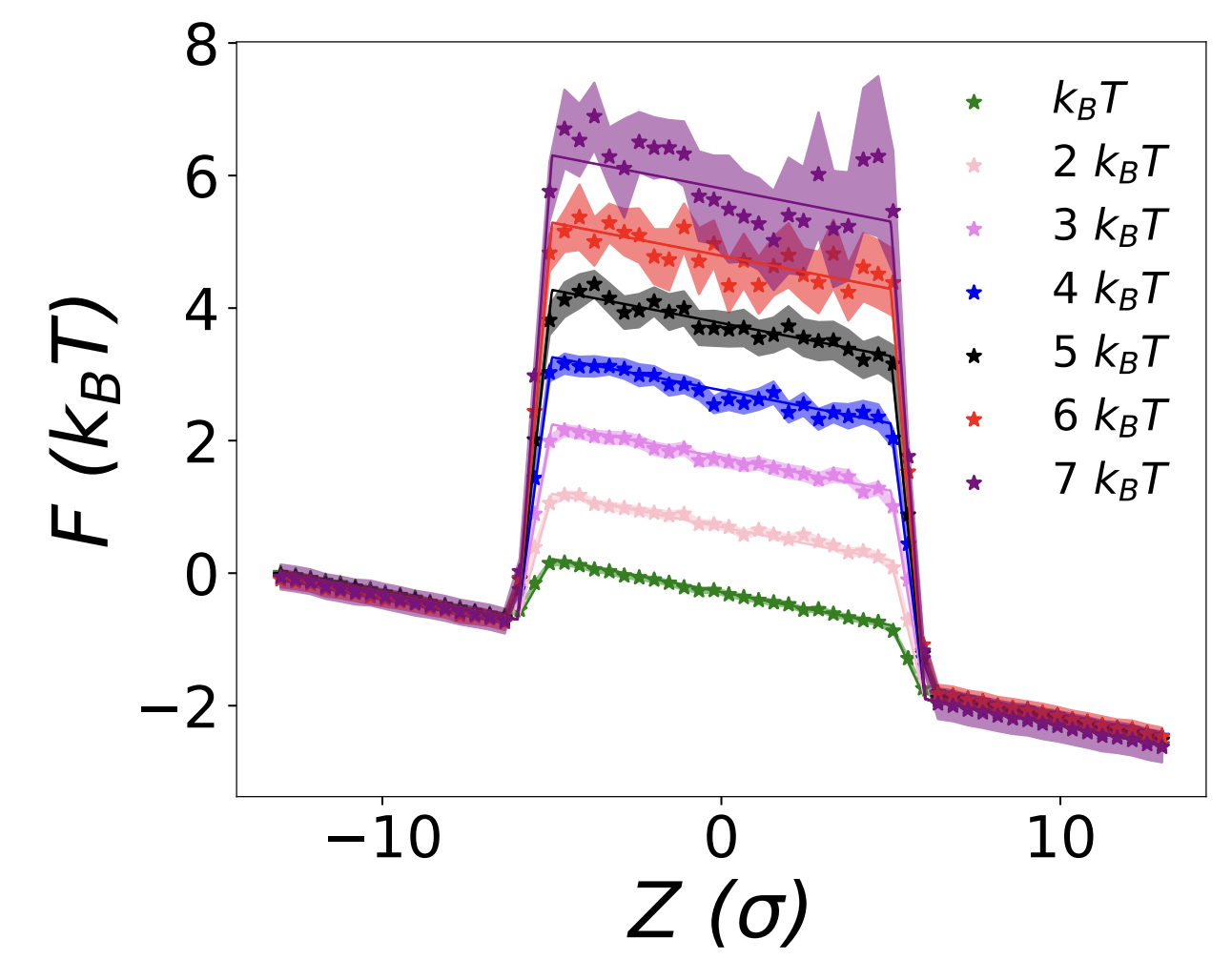
Non-equilibrium Trajectory Sampling (NETS) method for generating free-energy landscapes and steady-state distributions
Mohsen Farshad, Akwasi Nana Prempeh Ansah-Antwi, Pedro H. Amorim Valenca, Jonathan K. Whitmer
J. Chem. Phys. 163, 224125 (2025) DOI: 10.1063/5.0290883
This study presents a novel method for constructing free-energy profiles and steady-state distributions from either equilibrium or non-equilibrium trajectories along a defined reaction coordinate. The method works by tracking the final states of a swarm of short simulations launched under different initial conditions with no prior knowledge of the free energy landscape. Subsequently, this trajectory information is used to build a transition matrix whose primary eigenvector captures the steady-state occupation probability for each value of the reaction coordinate, yielding the free energy profile in equilibrium. This innovative method holds potential for many new materials and engineering applications where it is desired to know the free energy of rate-limiting configurations as may be relevant for transport processes (in, e.g., battery electrolytes and nano-filtration membranes), complexation (in, e.g., self-assembly and ligand-binding interactions), or in tuning properties such as adsorption. We illustrate the effectiveness of the method by capturing the free energy associated with a one-dimensional barrier potential modeling a separation membrane and the particle distribution associated with thermophoresis under a temperature gradient. Further extensions and applications of the method are also discussed.
A single-GPU implementation of first-principles molecular dynamics
François Gygi
J. Chem. Phys. 163, 154502 (2025). DOI: 10.1063/5.0288397
We present a single-Graphics Processing Unit (GPU) implementation of First-Principles Molecular Dynamics (FPMD) using plane wave basis functions and pseudopotentials, for the NVIDIA CUDA platform. We discuss various design choices made to exploit the high memory bandwidth available on a GPU while minimizing host–device data transfers. Applications to FPMD simulations of superionic NH3, liquid water, defects in SiC and MgO, and various systems including up to 512 atoms and 4096 electrons are demonstrated on an NVIDIA A100 GPU and an NVIDIA Grace-Hopper GH200 platform. Performance benchmarks show a significant speedup compared to other GPU-enhanced implementations, enabling the efficient use of computational resources. Applications to ensemble FPMD simulations of free energy barriers of a solvated molecule and residual stress in liquid water are also presented.
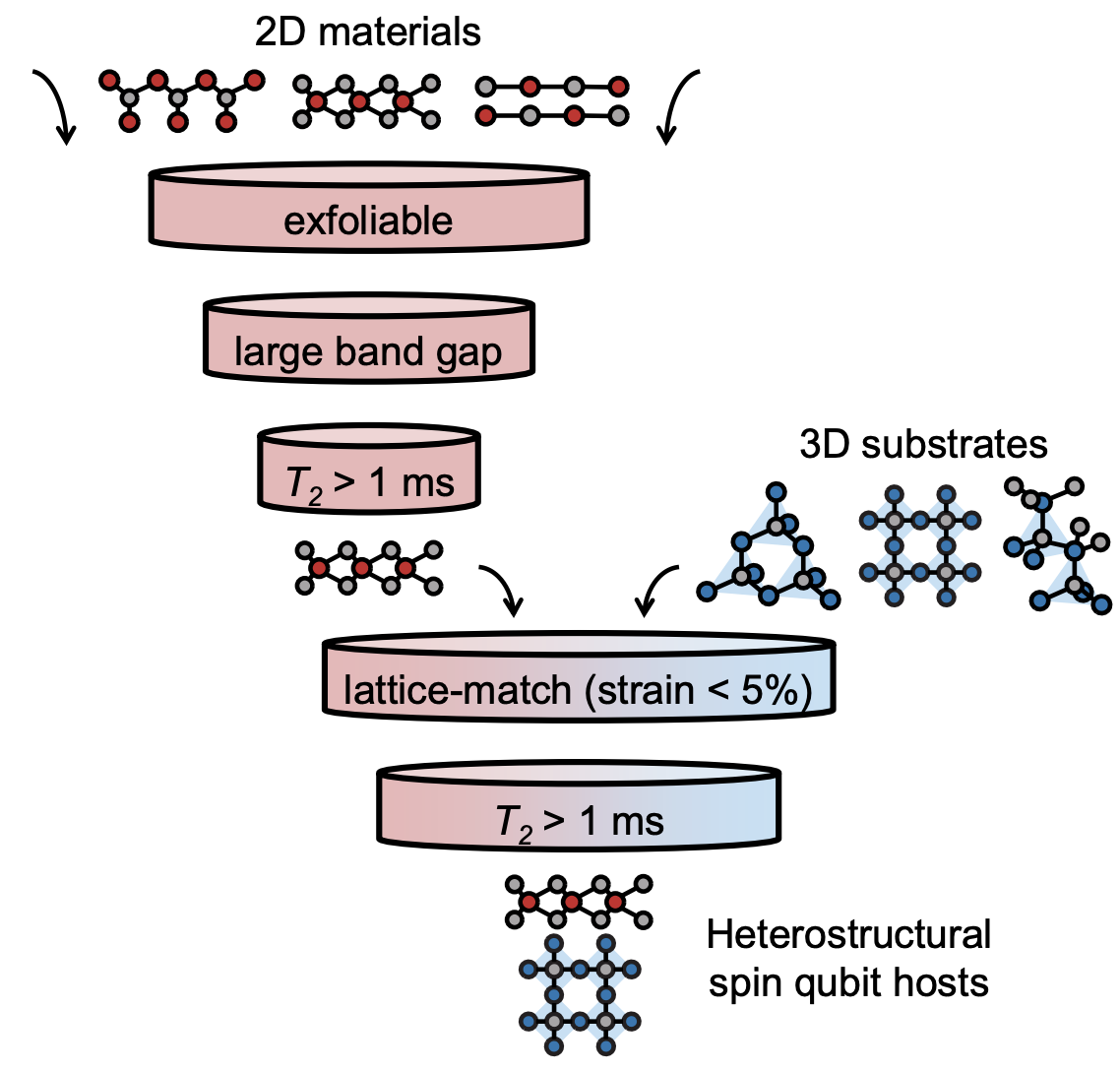
Strategies to search for two-dimensional materials with long spin qubit coherence time
Michael Y. Toriyama, Jiawei Zhan, Shun Kanai, Giulia Galli
npj 2D Mater. Appl. 9, 108 (2025). DOI: 10.1038/s41699-025-00623-8
Two-dimensional (2D) materials that can host qubits with long spin coherence time (T2) have the distinct advantage of integrating easily with existing microelectronic and photonic platforms, making them attractive for designing novel quantum devices with enhanced performance. However, the relative lack of 2D materials as spin qubit hosts, as well as appropriate substrates that can help maintain long T2, necessitates a strategy to search for candidates with robust spin coherence. Here, we develop a high-throughput computational workflow to predict the nuclear spin bath-driven qubit decoherence and T2 in 2D materials and heterostructures. We initially screen 1172 2D materials and find 189 monolayers with T2 > 1 ms, higher than that of naturally-abundant diamond. We then construct 1554 lattice-commensurate heterostructures between high-T2 2D materials and select 3D substrates, and we find that T2 is generally lower in a heterostructure than in the bare 2D host material; however, low-noise substrates (such as CeO2 and CaO) can help maintain high T2. To further accelerate the material screening effort, we derive analytical models that enable rapid predictions of T2 for 2D materials and heterostructures. The models offer a simple, yet quantitative, way to determine the relative contributions to decoherence from the nuclear spin baths of the 2D host and substrate in a heterostructural system. By developing a high-throughput workflow and analytical models, we expand the genome of 2D materials and their spin coherence times for the development of spin qubit platforms.

Many-body perturbation theory with hybrid density functional theory starting points accelerated by adaptively compressed exchange
Victor Wen-zhe Yu, Marco Govoni
J. Chem. Phys. 163, 024111 (2025). DOI: 10.1063/5.0239984
We report on the use of the adaptively compressed exchange (ACE) operator to accelerate many-body perturbation theory (MBPT) calculations, including G0W0 and the Bethe–Salpeter equation (BSE), for hybrid density functional theory starting points. We show that by approximating the exact exchange operator with the low-rank ACE operator, substantial computational savings can be achieved with systematically controllable errors in the quasiparticle energies computed with full-frequency G0W0 and the optical absorption spectra and vertical excitation energies computed by solving the BSE within density matrix perturbation theory. Our implementation makes use of the ACE-accelerated electronic Hamiltonian to carry out both G0W0 and BSE without explicitly computing empty states. We show the robustness of the approach and present the computational gains obtained on both the central processing unit and graphics processing unit nodes. Our work will facilitate the exploration and evaluation of fine-tuned hybrid starting points aimed at enhancing the accuracy of MBPT calculations without involving computationally demanding self-consistency in Hedin’s equations.
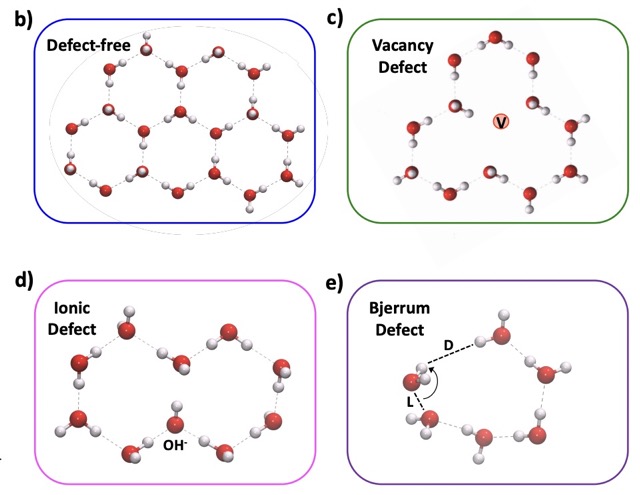
Defects at Play: Shaping the Photophysics and Photochemistry of Ice
Marta Monti, Yu Jin, Gonzalo Díaz Mirón, Arpan Kundu, Marco Govoni, Giulia Galli, Ali Hassanali
PNAS 122 (47) e2516805122, 2025. DOI: 10.1073/pnas.2516805122
The mechanisms by which light interacts with ice and the impact of photoinduced reactions are central to our understanding of environmental, atmospheric, and astrophysical processes. However, a microscopic description of the photoproducts originating from ultraviolet (UV) absorption and emission processes has remained elusive. Here, we explore the photochemistry of ice using time-dependent hybrid density functional theory on various models of pristine and defective ice Ih. Our investigation of the excited state potential energy surface of the crystal shows that UV absorption can lead to the formation of hydronium ions, hydroxyl radicals, and excess electrons. One of the dominant mechanisms of decay from the excited to the ground-state involves the recombination of the electron with the hydroxyl radical yielding hydronium-hydroxide ion-pairs. We find that the details of this charge recombination process sensitively depend on the presence of defects in the lattice, such as vacancies and preexisting photoproducts. We also observe the formation of Bjerrum defects following UV absorption; we suggest that, together with hydroxide anions, they are likely responsible for prominent features experimentally detected in long UV exposure absorption spectra, remarkably red-shifted relative to short exposure spectra. Our results highlight the key role of defects in determining the onset of absorption and emission processes in ice.
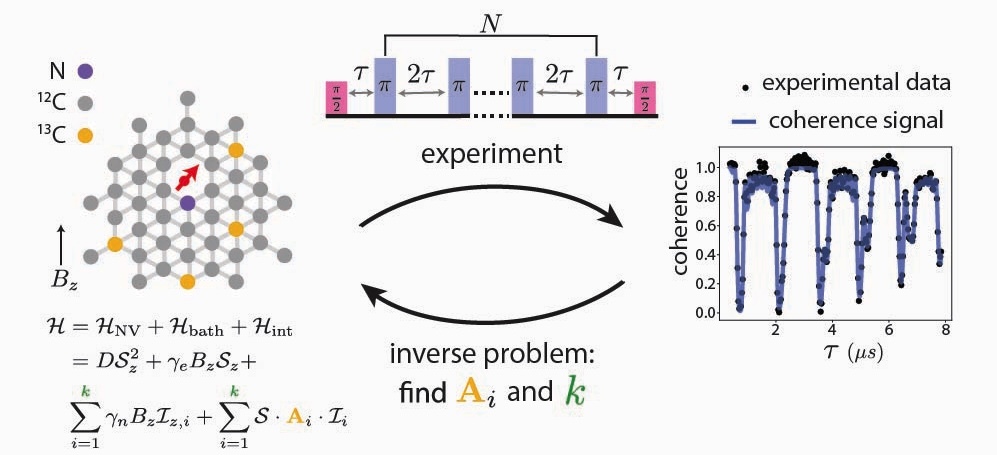
High-throughput spin-bath characterization of spin-defects in semiconductors
Abigail N. Poteshman, Mykyta Onizhuk, Christopher Egerstrom, Daniel P. Mark, David D. Awschalom, F. Joseph Heremans, Giulia Galli
Phys. Rev. Appl. 24, 054048 (2025). DOI: 10.1103/p57x-8kk7
Detailed knowledge of the local environments of spin-defects in semiconductors, such as nitrogen vacancy (NV) centers in diamond or divacancies in silicon carbide, is crucial for optimizing control and entanglement protocols in quantum sensing and information applications. However, a direct experimental characterization of individual defect environments is not scalable, as spin bath measurements are extremely time consuming. In this work, we address the ill-posed inverse problem of recovering the atomic positions and hyperfine couplings of random nuclei surrounding spin-defects from sparse experimental coherence signals, which can be obtained in hours. To address the challenge to determine the number of isotopic nuclear spins along with their hyperfine couplings, we employ a trans-dimensional Bayesian approach that incorporates ab initio data. This approach provides posterior distributions of the numbers, hyperfine couplings, and locations of nuclear spins present in the sample. In addition to enabling high-throughput screening of spin-defects, we demonstrate how this trans-dimensional Bayesian approach can guide experimental design for dynamical decoupling experiments to detect nuclear spins within targeted hyperfine coupling regimes. While the primary focus is on accelerating spin-defect characterization, this Bayesian approach also lays the foundation for digital twin studies of spin-defects, where a virtual model of the spin-defect system evolves in real time with ongoing experimental measurements. Together, the set of tools we designed and applied paves the way for scalable deployment of spin-defects in semiconductors for quantum sensing and information applications.

Autodifferentiable Geometric Restraints for Enhanced Sampling Simulations with Classical and Machine Learned Force Fields
Gustavo R. Pérez-Lemus, Cintia A. Menendez, Yinan Xu, Pablo F. Zubieta Rico, Yezhi Jin, Juan J. de Pablo
Submitted (2025). DOI: 10.48550/arXiv.2504.13575
The use of external restraints is ubiquitous in advanced molecular simulation techniques. In general, restraints serve to reduce the configurational space that is available for sampling, thereby reducing the computational demands associated with a given simulations. Examples include the use of positional restraints in docking simulations or positional restraints in studies of catalysis. Past work has sought to couple complex restraining potentials with enhanced sampling methods, including Metadynamics or Extended Adaptive Biasing Force approaches. Here, we introduce the use of more general geometric potentials coupled with enhanced sampling methods that incorporate neural networks or spectral decomposition to achieve more efficient sampling in the context of advanced materials design.

Considerations in the use of machine learning force fields for free energy calculations
Orlando A. Mendible, Jonathan K. Whitmer, Yamil J. Colón
J. Chem. Phys. 162, 174119 (2025). DOI: 10.1063/5.0252043
Machine learning force fields (MLFFs) promise to accurately describe the potential energy surface of molecules at the ab initio level of theory with improved computational efficiency. Within MLFFs, equivariant graph neural networks (EQNNs) have shown great promise in accuracy and performance and are the focus of this work. The capability of EQNNs to recover free energy surfaces (FES) remains to be thoroughly investigated. In this work, we investigate the impact of collective variables (CVs) distribution within the training data on the accuracy of EQNNs predicting the FES of butane and alanine dipeptide. A generalizable workflow is presented in which training configurations are generated with classical molecular dynamics simulations, and energies and forces are obtained with ab initio calculations. We evaluate how bond and angle constraints in the training data influence the accuracy of EQNN force fields in reproducing the FES of the molecules at both classical and ab initio levels of theory. Results indicate that the model’s accuracy is unaffected by the distribution of sampled CVs during training, given that the training data includes configurations from characteristic regions of the system’s FES. However, when the training data is obtained from classical simulations, the EQNN struggles to extrapolate the free energy for configurations with high free energy. In contrast, models trained with the same configurations on ab initio data show improved extrapolation accuracy. The findings underscore the difficulties in creating a comprehensive training dataset for EQNNs to predict FESs and highlight the importance of prior knowledge of the system’s FES.

Long Molecular Wires and the Auto-ionization of Water
Yinan Xu, Samuel Varner, Yezhi Jin, Gustavo Pérez-Lemus, Joan Montes de Oca, Paul Nealey, Seth Darling, Zhen-Gang Wang, Juan de Pablo
Submitted (2025). DOI: 10.26434/chemrxiv-2024-f9bv7-v2
Water auto-ionization is critical in a wide range of chemical, biological, physical, and industrial processes. In this work, we describe a series of hitherto unknown collective molecular processes leading to auto-ionization. Specifically, by combining machine-learned interatomic potentials and spectral adaptive biasing force techniques, we determine the relevant free energy landscape of water auto-ionization. At ambient conditions, the free energy profile reveals two distinct saddle points, each leading to the formation of three- and four-member water wires. The wires feature an individual Zundel ion and a proton diffusion-like transition state, respectively. At elevated temperatures, the auto-ionization process exhibits a more concerted hydrogen transfer mechanism and reveals an alternative pathway involving the synchronous diffusion of Zundel ion pairs, with the ion pair corresponding to an energetic local minimum on the free energy surface. These findings help resolve long-standing conflicting views of the mechanism of water auto-ionization and provide new avenues for the study of proton behavior in different aqueous environments.

Free-Energy Landscapes and Surface Dynamics in Methane Activation on Ni(511) via Machine Learning and Enhanced Sampling
Yezhi Jin, Yinan Xu, Jireh Garcia Sanchez, Gustavo Perez-Lemus, Pablo Zubieta Rico, Massimiliano Delferro, Juan de Pablo
ACS Catal. 15, 11, 8931–8942 (2025). DOI: 10.1021/acscatal.5c00724
Methane activation on stepped Ni(511) surfaces involves the rearrangement of surface atoms as the chemical reaction proceeds. This process is particularly sensitive to temperature. Using machine-learned interatomic potentials (MLIPs) coupled with enhanced sampling techniques, we investigate the activation of methane under realistic operando conditions. Our analysis reveals that methane dissociation occurs predominantly at step-edge nickel atoms. As CHx (where x = 3 or 4) species bind to additional surface nickel atoms, their reduced mobility leads to entropic penalties that suppress certain configurations and transition states. This is reflected in the underlying free energy surfaces, where configurations such as methyl binding to hollow sites and activation routes involving two nickel atoms become unfavorable as temperature increases. At elevated temperatures, methane activation extends from step-edge sites to terrace regions because of reduced free-energy barriers and enhanced surface dynamics. By decomposing the free-energy into enthalpic and entropic contributions, we uncover temperature-dependent shifts in the preferences of methane for the relevant active sites and arrive at a detailed molecular picture of methane activation.

Designing Optically Addressable Nitrogen-Vacancy Centers in Ultrasmall Nanodiamonds: Insights from First-Principles Calculations
Arpan Kundu*, Francesco Martinelli*, and Giulia Galli (*equal contribution)
J. Phys. Chem. Lett. 16 (8), 1973-1979 (2025). DOI: 10.1021/acs.jpclett.5c00355
Ultrasmall nanodiamonds (USNDs) are promising platforms for fluorescent and quantum-sensing applications. Here we present first-principles electronic structure calculations of nitrogen-vacancy (NV-) centers in USNDs, and we investigate their optical addressability as a function of the surface termination. We consider both isolated nanoparticles and arrays of USNDs with different degrees of packing, and we include quantum vibronic effects in our analysis, using stochastic methods. We find that the NV- can be stabilized in a negative charge state if the nanoparticles are terminated by fluorine, hydroxyl, and ether. While fluorine terminations are useful for fluorescent biotags, we suggest that hydroxyl and ether terminations are beneficial for quantum-sensing applications. We also find that the NV- can be stabilized in arrays of USNDs for interparticle separations larger than the diameter of the nanoparticle. Finally, our results show that in arrays, electron phonon interactions enhance the negative charge stability of NV- centers.

Strongly Correlated States of Transition Metal Spin Defects: The Case of an Iron Impurity in Aluminum Nitride
Leon Otis, Yu Jin, Victor Wen-zhe Yu, Siyuan Chen, Laura Gagliardi*, and Giulia Galli*
J. Phys. Chem. Lett. 16 (12), 3092-3099 (2025). DOI: 10.1021/acs.jpclett.5c00287
We investigate the electronic properties of an exemplar transition metal impurity in an insulator, with the goal of accurately describing strongly correlated defect states. We consider iron in aluminum nitride, a material of interest for hybrid quantum technologies, and we carry out calculations with quantum embedding methods, density matrix embedding theory (DMET) and quantum defect embedding theory (QDET), and with spin-flip time-dependent density functional theory (TDDFT). We show that both DMET and QDET accurately describe the ground state and low-lying excited states of the defect and that TDDFT yields photoluminescence spectra in agreement with experiments. In addition, we provide a detailed discussion of the convergence of our results as a function of the active space used in the embedding methods, thus defining a protocol to obtain converged data directly comparable with experiments.

An NV− center in magnesium oxide as a spin qubit for hybrid quantum technologies
Vrindaa Somjit, Joel Davidsson, Yu Jin & Giulia Galli
npj Comput Mater 11, 74 (2025). DOI: 10.1038/s41524-025-01558-w
Recent predictions suggest that oxides, such as MgO and CaO, could serve as hosts of spin defects with long coherence times and thus be promising materials for quantum applications. However, in most cases, specific defects have not yet been identified. Here, by using a high-throughput first-principles framework and advanced electronic structure methods, we identify a negatively charged complex between a nitrogen interstitial and a magnesium vacancy in MgO with favorable electronic and optical properties for hybrid quantum technologies. We show that this NV− center has stable triplet ground and excited states, with singlet shelving states enabling optical initialization and spin-dependent readout. We predict several properties, including absorption, emission, and zero-phonon line energies, as well as zero-field splitting tensor, and hyperfine interaction parameters, which can aid in the experimental identification of this defect. Our calculations show that due to a strong pseudo-Jahn Teller effect and low-frequency phonon modes, the NV− center in MgO is subject to a substantial vibronic coupling. We discuss design strategies to reduce such coupling and increase the Debye-Waller factor, including the effect of strain and the localization of the defect states. We propose that the favorable properties of the NV− defect, along with the technological maturity of MgO, could enable hybrid classical-quantum applications, such as spintronic quantum sensors and single qubit gates.

Connectivity-dependent Exciton-phonon Coupling in Cesium Bismuth Halide Quantum Dots
Beiye Li, Hugh Cairney, Yu Jin, Jinsoo Park, Siddhartha Sohoni, Lawson Lloyd, Yuzi Liu, Justin Jureller, Young Jay Ryu, Stella Chariton, Vitali Prakapenka, Richard Schaller, Giulia Galli, and Gregory Engel
ACS Nano 19 (10), 10359-10368 (2025). DOI: 10.1021/acsnano.4c18414
Metal halide octahedra form the fundamental functional building blocks of metal halide perovskites, dictating their structures, optical properties, electronic structures, and dynamics. In this study, we show that the connectivity of bismuth halide octahedra in Cs3Bi2Br9 and Cs3Bi2I9 quantum dots (QDs) changes with different halide elements. We use first-principles calculations to reveal the key role of the connectivity of bismuth halide octahedra on the wave function symmetry, Huang–Rhys factor, and exciton–phonon interaction strength. Following QD synthesis via a ligand-mediated transport method, the effect of connectivity is verified with transient absorption spectroscopy, where we contrast Cs3Bi2Br9 and Cs3Bi2I9 QD exciton dynamics. In photoexcited Cs3Bi2I9 QDs, phonons related to the vibrational motions of face-sharing [BiI6]3– bioctahedra couple strongly to the electronic state and drive rapid carrier relaxation. Equivalent signals are not observed for photoexcited Cs3Bi2Br9 QDs, implying a lack of phonon involvement in band-edge absorption and subsequent exciton relaxation. Our findings suggest that structural engineering can effectively tune the exciton–phonon coupling and therefore influence exciton relaxation and recombination in perovskite nanomaterials.

Dielectric-Dependent Range-Separated Hybrid Functional Calculations for Metal Oxides
Jiawei Zhan, Marco Govoni, Giulia Galli
Phys. Rev. Materials 9, 053808 (2025). DOI: 10.1103/PhysRevMaterials.9.053808
Recently, we introduced the screened-exchange range-separated hybrid (SE-RSH) functional to account for spatially dependent dielectric screening in complex materials. The SE-RSH functional has shown good performance in predicting the electronic properties of a large variety of semiconductors and insulators, and of heterogeneous systems composed of building blocks with large dielectric mismatch. Here, we assess the performance of SE-RSH for oxide materials, including antiferromagnetic transition-metal oxides. Through a comparison with other dielectric-dependent hybrid functionals, we demonstrate that SE-RSH yields improved predictions of dielectric constants and band gaps, bringing them into a closer agreement with experimental values. The functional also provides accurate values of magnetic moments of several oxides.

First-Principles Framework for the Prediction of Intersystem Crossing Rates in Spin Defects: The Role of Electron Correlation
Yu Jin, Jinsoo Park, Marquis M. McMillan, Daniel Donghyon Ohm, Corrie Barnes, Benjamin Pingault, Christopher Egerstrom, Benchen Huang, Marco Govoni, F. Joseph Heremans, David D. Awschalom, and Giulia Galli
Phys. Rev. Lett. 135, 036401 (2025), Editor's Suggestion . DOI: 10.1103/nw3r-zy8q
Optically active spin defects in solids are promising platforms for quantum technologies. Here, we present a first-principles framework to investigate intersystem crossing processes, which represent crucial steps in the optical spin-polarization cycle used to address spin defects. Considering the nitrogen-vacancy center in diamond as a case study, we demonstrate that our framework effectively captures electron correlation effects in the calculation of many-body electronic states and their spin-orbit coupling and electron-phonon interactions, while systematically addressing finite-size effects. We validate our predictions by carrying out measurements of fluorescence lifetimes, finding excellent agreement between theory and experiments. The framework presented here provides a versatile and robust tool for exploring the optical cycle of varied spin defects entirely from first principles.

Computational study of indium oxide photoelectrodes
Matthew Bousquet, Jiawei Zhan, Chunxin Luo, Alex Martinson, Francois Gygi, and Giulia Galli
J. Phys. Chem. C 129, 17, 8395–8403 (2025). DOI: 10.1021/acs.jpcc.5c01077
Using a combination of first-principles molecular dynamics simulations and electronic structure calculations, we characterize the atomistic structure and vibrational properties of a photocatalytic surface of In2O3, a promising photoelectrode for the production of hydrogen peroxide. We then investigate the surface in contact with water and show that the electronic states of In2O3 are appropriately positioned in energy to facilitate the two-electron water oxidation reaction (WOR) over the competing four-electron oxygen evolution reaction. We further propose that the use of strained thin films interfaced with water is beneficial in decreasing the optical gap of In2O3 and thus in utilizing a wider portion of the solar spectrum for the WOR.

Advances in quantum defect embedding theory
Siyuan Chen, Victor Wen-zhe Yu, Yu Jin, Marco Govoni, and Giulia Galli
J. Chem. Theory Comput. 21, 16, 7797-7812 (2025) DOI: 10.1021/acs.jctc.5c00559
Quantum defect embedding theory (QDET) is a many-body embedding method designed to describe condensed systems with strongly correlated electrons localized within a given region of space, for example spin defects in semiconductors and insulators. Although the QDET approach has been successful in predicting the electronic properties of several point defects, several limitations of the method remain. In this work, we propose multiple advances to the QDET formalism. We derive a double-counting correction that consistently treats the frequency dependence of the screened Coulomb interaction, and we illustrate the effect of including unoccupied orbitals in the active space. In addition, we propose a method to describe hybridization effects between the active space and the environment, and we compare the results of several impurity solvers, providing further insights into improving the reliability and applicability of the method. We present results for defects in diamond and for molecular qubits, including a detailed comparison with experiments.

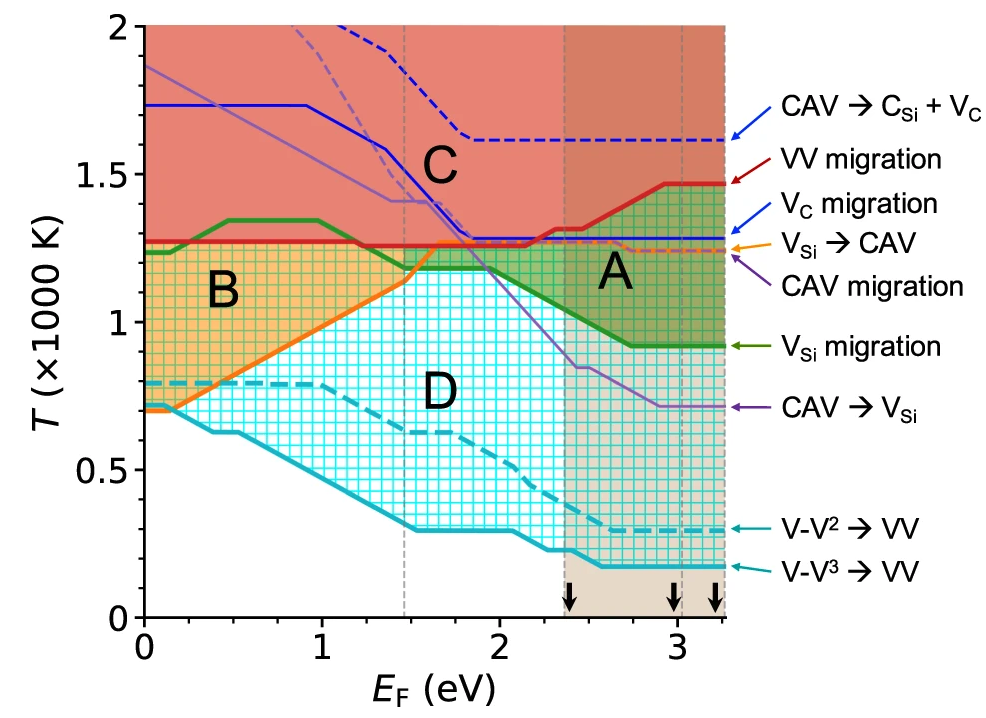
Computationally guided experimental validation of divacancy defect formation in 4H-SiC
Taishi Kimura, Jonghoon Ahn, Nazar Delegan, Alan Dibos, Jiefei Zhang, Benjamin Pingault, Cunzhi Zhang, Giulia Gali, David Awschalom, and F. Joseph Heremans
Appl. Phys. Lett. 126, 164001. (2025) DOI: 10.1063/5.0255575
Recent research into solid-state qubits for quantum information science has focused on optically addressable spin defects such as the negatively charged nitrogen-vacancy center in diamond and the neutrally charged divacancy (VV) in 4H-SiC as scalable quantum sensors and networking qubits. Within this context, direct investigations of the structural origin and defect formation dynamics of a sub-set of the VV center in 4H-SiC remain lacking. Here, we take a systematic experimental approach guided by predictions from first-principles simulations to gain a thorough mechanistic understanding of the VV defect formation and control in 4H-SiC. We study the effect of annealing time and temperature on VV formation in high-purity semi-insulating 4H-SiC samples following electron irradiation. Three different temperatures (1123, 1273, and 1473 K) and annealing duration (from 0.5 to 72 h) are chosen to explore VV formation in different regions. We find that samples annealed at 1273 K give the highest VV-related photoluminescence (PL) intensities, in agreement with the prediction from first-principles calculations. Furthermore, the logarithmic dependence of VV-related PL intensities on the annealing duration at 1273 K indicates that 1273 K provides sufficient thermal energy for silicon vacancy migration but not for VV migration. Together, these results suggest that efficient VV formation occurs above the VSi migration temperature and below the VV migration threshold.
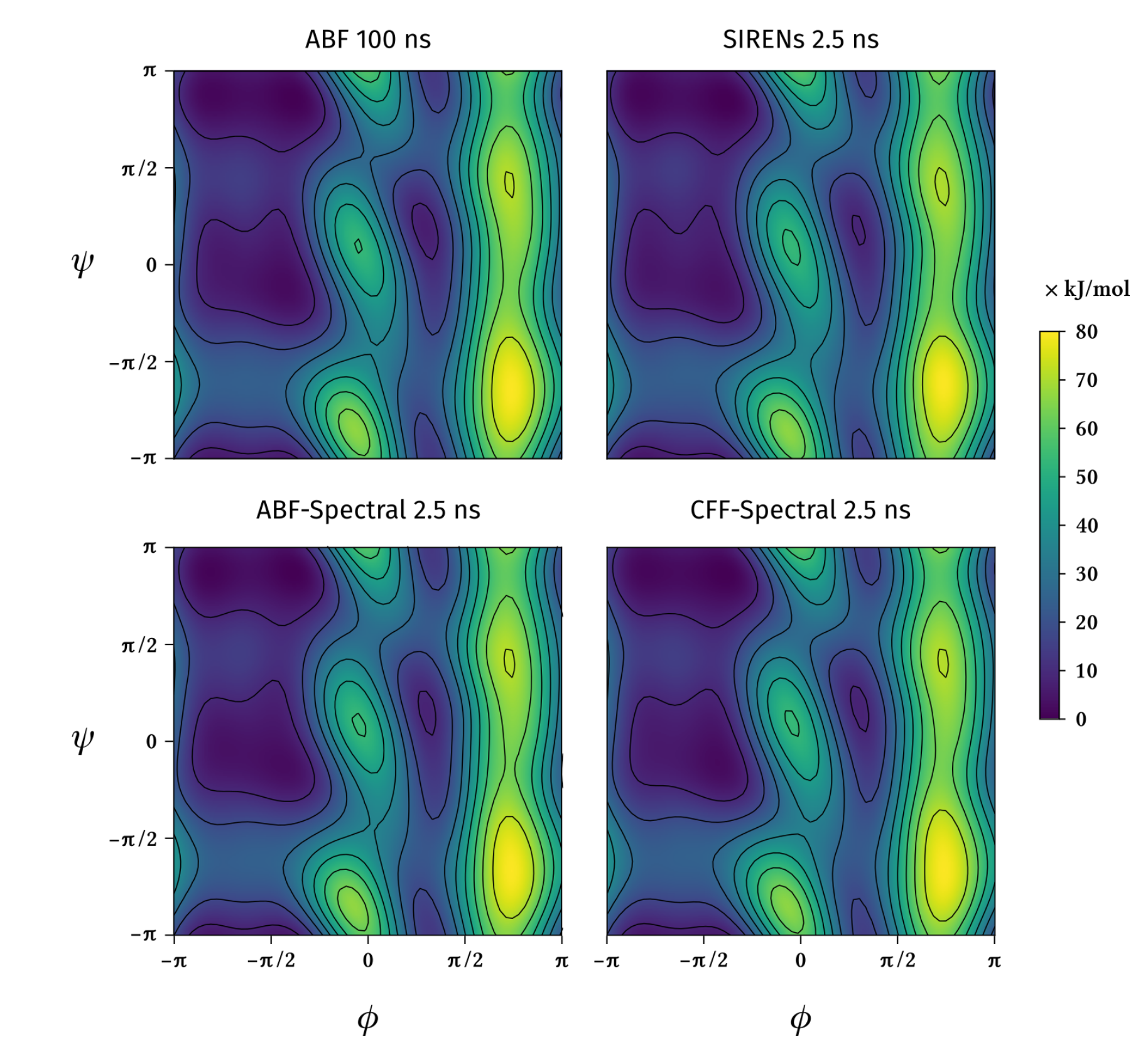
Efficient sampling of free energy landscapes with functions in Sobolev spaces
Pablo F. Zubieta Rico, Gustavo R. Pérez-Lemus, and Juan J. de Pablo
J. Chem. Phys. 162, 084109 (2025). DOI:10.1063/5.0221263
Molecular simulations of biological and physical phenomena generally involve sampling complicated, rough energy landscapes characterized by multiple local minima. In this work, we introduce a new family of methods for advanced sampling that draw inspiration from functional representations used in machine learning and approximation theory. As shown here, such representations are particularly well suited for learning free energies using artificial neural networks. As a system evolves through phase space, the proposed methods gradually build a model for the free energy as a function of one or more collective variables, from both the frequency of visits to distinct states and generalized force estimates corresponding to such states. Implementation of the methods is relatively simple and, more importantly, for the representative examples considered in this work, they provide computational efficiency gains of up to several orders of magnitude over other widely used simulation techniques.

The importance of sampling the dynamical modes: Reevaluating benchmarks for invariant and equivariant features of machine learning potentials for simulation of free energy landscapes
Gustavo Perez-Lemus, Yinan Xu, Yezhi Jin, Pablo Zubieta Rico, Juan de Pablo
J. Chem. Phys. 161, 244703 (2024). DOI: 10.1063/5.0237399
Machine learning interatomic potentials (MLIPs) are rapidly gaining interest for molecular modeling, as they provide a balance between quantum-mechanical level descriptions of atomic interactions and reasonable computational efficiency. However, questions remain regarding the stability of simulations using these potentials, as well as the extent to which the learned potential energy function can be extrapolated safely. Past studies have encountered challenges when MLIPs are applied to classical benchmark systems. In this work, we show that some of these challenges are related to the characteristics of the training datasets, particularly the inefficient exploration of the dynamical modes and the inclusion of rigid constraints. We demonstrate that long stability in simulations with MLIPs can be achieved by generating unconstrained datasets using unbiased classical simulations, provided that the important dynamical modes are correctly sampled. In addition, we emphasize that in order to achieve precise energy predictions, it is important to resort to enhanced sampling techniques for dataset generation, and we demonstrate that safe extrapolation of MLIPs depends on judicious choices related to the system’s underlying free energy landscape and the symmetry features embedded within the machine learning models.

A Molecular View of Methane Activation on Ni(111) through Enhanced Sampling and Machine Learning
Yinan Xu, Yezhi Jin, Jireh S. García Sánchez, Gustavo R. Pérez-Lemus, Pablo F. Zubieta Rico, Massimiliano Delferro, Juan J. de Pablo
J. Phys. Chem. Lett. 15, 39, 9852-9862 (2024). DOI: 10.1021/acs.jpclett.4c02237
A combination of machine learned interatomic potentials (MLIPs) and enhanced sampling simulations is used to investigate the activation of methane on a Ni(111) surface. The work entails the development and iterative refinement of MLIPs, initially trained on a data set constructed via ab initio molecular dynamics simulations, supplemented by adaptive biasing forces, to enrich the sampling of catalytically relevant configurations. Our results reveal that upon incorporation of collective variables that capture the behavior of the reactant molecule, as well as additional frames that describe the dynamic response of the catalytic surface, it is possible to enhance considerably the accuracy of predicted energies and forces. By employing enhanced sampling schemes in the refinement of the MLIP, we systematically explore the potential energy surface, leading to a refined MLIP capable of predicting density functional theory-level energies and forces and replicating key geometric characteristics of the catalytic system. The resulting free energy landscapes at several temperatures provide a detailed view of the thermodynamics and dynamics of methane activation. Specifically, as methane approaches and dissociates on the catalytic surface, the process involves the dynamic interplay of CH4 and the Ni catalyst that includes both enthalpic and entropic contributions. The progression toward the transition state involves a CH4 moiety that is increasingly restrained in its ability to rotate or translate, while the stage following the transition state is characterized by a notable rise of the Ni atom that interacts with the cleaved C–H bond. This leads to an increase in the mobility of the adsorbed species, a feature that becomes more pronounced at higher temperatures.

Polarization-dependent photoluminescence of Ce-implanted MgO and MgAl2O4
Manato Kawahara, Yuichiro Abe, Koki Takano, F. Joseph Heremans, Jun Ishihara, Sean E.Sullivan, Christian Vorwerk, Vrindaa Somjit, Christopher P.Anderson, Gary Wolfowicz, Makoto Kohda, Shunsuke Fukami, Giulia Galli, David D. Awschalom, Hideo Ohno, and Shun Kanai.
Appl. Phys. Express 17, 072004 (2024). 10.35848/1882-0786/ad59f4
Since the qubit's performance of solid-state spin centers depends highly on the host material, spin centers using new host materials may offer new qubit applications. We investigate the optical properties of Ce-implanted MgO and MgAl2O4 as potential materials holding the optically accessible qubit. We find that the photoluminescence of Ce-implanted MgAl2O4 is more than 10 times brighter than that of Ce-implanted MgO and observe polarization-dependent emission of Ce center in MgAl2O4 with 2% at 4 K under 500 mT, suggesting that the properties required for initializing and reading the state of the spin qubit have been achieved.

Quantum Spin Probe of Single Charge Dynamics
Jonathan C. Marcks, Mykyta Onizhuk, Yu-Xin Wang, Benjamin Soloway, Masaya Fukami, Nazar Delegan, F. Joseph Heremans, Aashish A. Clerk, Giulia Galli, and David D. Awschalom
Phys. Rev. Lett. 133, 130802 (2024). 10.1103/PhysRevLett.133.130802
Electronic defects in semiconductors form the basis for emerging quantum technologies, but many defect centers are difficult to access at the single-particle level. A method for probing optically inactive spin defects would reveal semiconductor physics at the atomic scale and advance the study of new quantum systems. We exploit the intrinsic correlation between the charge and spin states of defect centers to measure the charge populations and dynamics of single substitutional nitrogen spin defects in diamond. By probing their steady-state spin population, read out at the single-defect level with a nearby nitrogen vacancy center, we directly measure the defect ionization—corroborated by first-principles calculations—an effect we do not have access to with traditional coherence-based quantum sensing.

Coherent Erbium Spin Defects in Colloidal Nanocrystal Hosts
Joeson Wong, Mykyta Onizhuk, Jonah Nagura, Arashdeep Singh Thind, Jasleen Bindra, Christina Wicker, Gregory Grant, Yuxuan Zhang, Jens Niklas, Oleg Poluektov, Robert Klie, Jiefei Zhang, Giulia Galli, F. Joseph Heremans, David Awschalom, and A. Paul Alivisatos
ACS Nano 18, 29, 19110–19123 (2024) DOI:10.1021/acsnano.4c04083
We demonstrate nearly a microsecond of spin coherence in Er3+ ions doped in cerium dioxide nanocrystal hosts, despite a large gyromagnetic ratio and nanometric proximity of the spin defect to the nanocrystal surface. The long spin coherence is enabled by reducing the dopant density below the instantaneous diffusion limit in a nuclear spin-free host material, reaching the limit of a single erbium spin defect per nanocrystal. We observe a large Orbach energy in a highly symmetric cubic site, further protecting the coherence in a qubit that would otherwise rapidly decohere. Spatially correlated electron spectroscopy measurements reveal the presence of Ce3+ at the nanocrystal surface, which likely acts as extraneous paramagnetic spin noise. Even with these factors, defect-embedded nanocrystal hosts show tremendous promise for quantum sensing and quantum communication applications, with multiple avenues, including core–shell fabrication, redox tuning of oxygen vacancies, and organic surfactant modification, available to further enhance their spin coherence and functionality in the future.
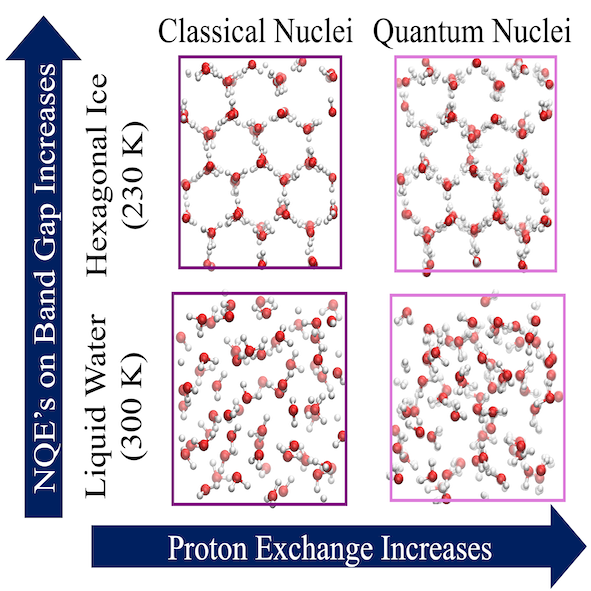
Nuclear Quantum Effects on the Electronic Structure of Water and Ice
Margaret L. Berrens, Arpan Kundu, Marcos F. Calegari Andrade, Tuan Anh Pham, Giulia Galli, and Davide Donadio
J. Phys.Chem. Lett. 15, 6818-6825 (2024) DOI: 10.1021/acs.jpclett.4c01315
The electronic properties and optical response of ice and water are intricately shaped by their molecular structure, including the quantum mechanical nature of the hydrogen atoms. Despite numerous previous studies, a comprehensive understanding of the nuclear quantum effects (NQEs) on the electronic structure of water and ice at finite temperatures remains elusive. Here, we utilize molecular simulations that harness efficient machine-learning potentials and many-body perturbation theory to assess how NQEs impact the electronic bands of water and hexagonal ice. By comparing path-integral and classical simulations, we find that NQEs lead to a larger renormalization of the fundamental gap of ice, compared to that of water, ultimately yielding similar bandgaps in the two systems, consistent with experimental estimates. Our calculations suggest that the increased quantum mechanical delocalization of protons in ice, relative to water, is a key factor leading to the enhancement of NQEs on the electronic structure of ice.

GPU-Accelerated Solution of the Bethe-Salpeter Equation for Large and Heterogeneous Systems
Victor Wen-zhe Yu, Yu Jin, Giulia Galli, and Marco Govoni
J. Chem. Theory Comput. 20, 24, 10899-10911 (2024) DOI: 10.1021/acs.jctc.4c01253
We present a massively parallel GPU-accelerated implementation of the Bethe–Salpeter equation (BSE) for the calculation of the vertical excitation energies (VEEs) and optical absorption spectra of condensed and molecular systems, starting from single-particle eigenvalues and eigenvectors obtained with density functional theory. The algorithms adopted here circumvent the slowly converging sums over empty and occupied states and the inversion of large dielectric matrices through a density matrix perturbation theory approach and a low-rank decomposition of the screened Coulomb interaction, respectively. Further computational savings are achieved by exploiting the nearsightedness of the density matrix of semiconductors and insulators to reduce the number of screened Coulomb integrals. We scale our calculations to thousands of GPUs with a hierarchical loop and data distribution strategy. The efficacy of our method is demonstrated by computing the VEEs of several spin defects in wide-band-gap materials, showing that supercells with up to 1000 atoms are necessary to obtain converged results. We discuss the validity of the common approximation that solves the BSE with truncated sums over empty and occupied states. We then apply our GW-BSE implementation to a diamond lattice with 1727 atoms to study the symmetry breaking of triplet states caused by the interaction of a point defect with an extended line defect.
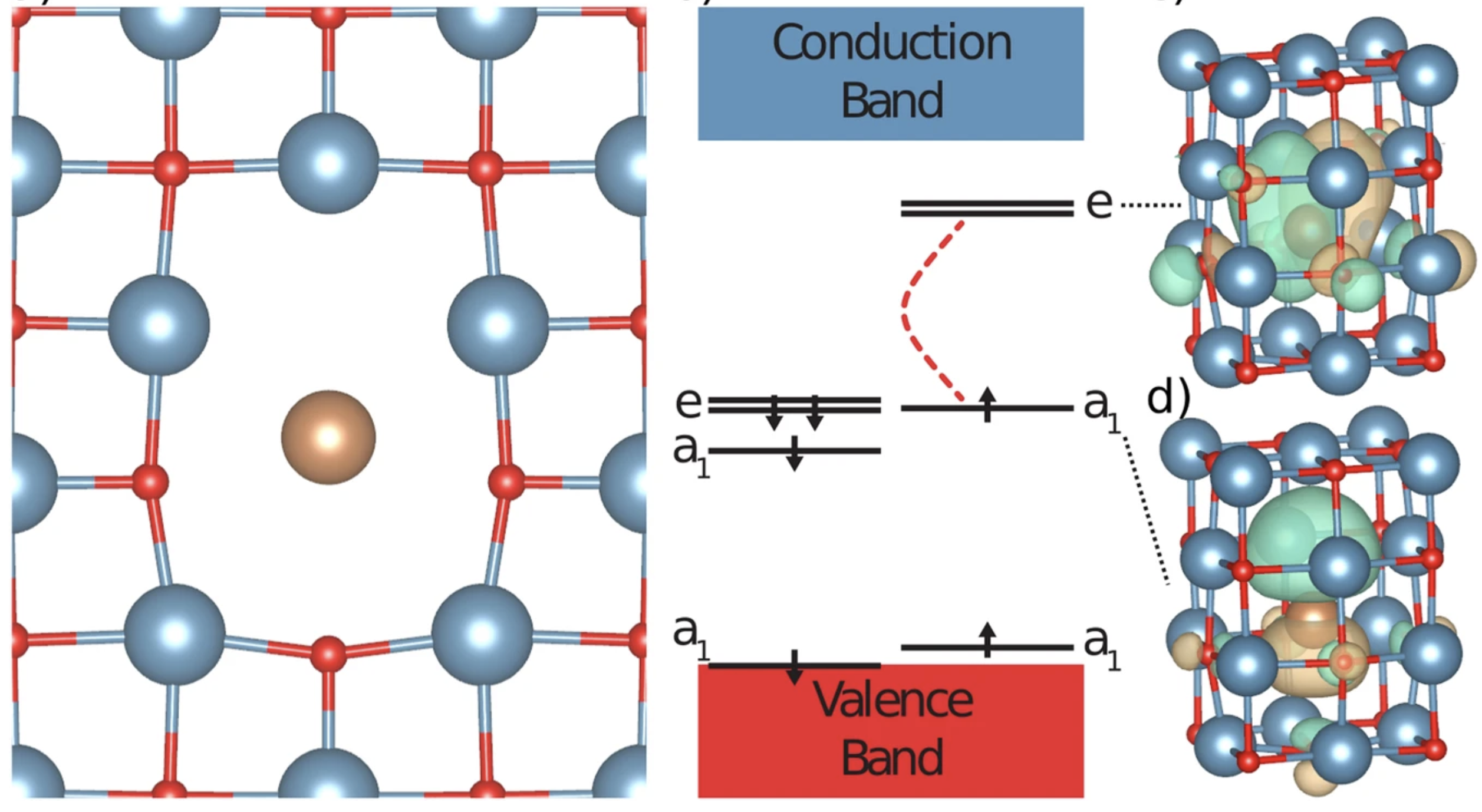
Discovery of Atomic Clock-Like Spin Defects in Simple Oxides from First Principles
Joel Davidsson, Mykyta Onizhuk, Christian Vorwerk, and Giulia Galli
Nat. Commun., 15, 4812 (2024) DOI: 10.1038/s41467-024-49057-8
Virtually noiseless due to the scarcity of spinful nuclei in the lattice, simple oxides hold promise as hosts of solid-state spin qubits. However, no suitable spin defect has yet been found in these systems. Using high-throughput first-principles calculations, we predict spin defects in calcium oxide with electronic properties remarkably similar to those of the NV center in diamond. These defects are charged complexes where a dopant atom — Sb, Bi, or I — occupies the volume vacated by adjacent cation and anion vacancies. The predicted zero phonon line shows that the Bi complex emits in the telecommunication range, and the computed many-body energy levels suggest a viable optical cycle required for qubit initialization. Notably, the high-spin nucleus of each dopant strongly couples to the electron spin, leading to many controllable quantum levels and the emergence of atomic clock-like transitions that are well protected from environmental noise. Specifically, the Hanh-echo coherence time increases beyond seconds at the clock-like transition in the defect with 209Bi. Our results pave the way to designing quantum states with long coherence times in simple oxides, making them attractive platforms for quantum technologies.

Charge state and entropic effects affecting the formation and dynamics of divacancies in 3C-SiC
Cunzhi Zhang, Francois Gygi, and Giulia Galli
Phys. Rev. Mater, 8, 046201 (2024) DOI: 10.1103/PhysRevMaterials.8.046201
Using nudged elastic band calculations and first-principles molecular dynamics with enhanced sampling, we study the formation and dynamics of the divacancy (VV) in cubic silicon carbide, including VV rotation and migration. We show that for all processes studied here the energy barriers and preferred pathway depend on the charge state of the defects. Our results indicate that the influence of multiple charge states and entropic effects should be considered for a quantitative description of the physical and dynamical properties of point defects at finite temperatures. In addition, we demonstrate that molecular dynamics simulations using machine-learning potentials can efficiently and reliably capture entropic effects and yield accurate free-energy barriers.

PySAGES: flexible, advanced sampling methods accelerated with GPUs
Pablo F. Zubieta Rico, Ludwig Schneider, Gustavo R. Pérez-Lemus, Riccardo Alessandri, Siva Dasetty, Trung D. Nguyen, Cintia A. Menéndez, Yiheng Wu, Yezhi Jin, Yinan Xu, Samuel Varner, John A. Parker, Andrew L. Ferguson, Jonathan K. Whitmer & Juan J. de Pablo
npj Computational Materials, 10, 35 (2024) DOI: 10.1038/s41524-023-01189-z
Molecular simulations are an important tool for research in physics, chemistry, and biology. The capabilities of simulations can be greatly expanded by providing access to advanced sampling methods and techniques that permit calculation of the relevant underlying free energy landscapes. In this sense, software that can be seamlessly adapted to a broad range of complex systems is essential. Building on past efforts to provide open-source community-supported software for advanced sampling, we introduce PySAGES, a Python implementation of the Software Suite for Advanced General Ensemble Simulations (SSAGES) that provides full GPU support for massively parallel applications of enhanced sampling methods such as adaptive biasing forces, harmonic bias, or forward flux sampling in the context of molecular dynamics simulations. By providing an intuitive interface that facilitates the management of a system’s configuration, the inclusion of new collective variables, and the implementation of sophisticated free energy-based sampling methods, the PySAGES library serves as a general platform for the development and implementation of emerging simulation techniques. The capabilities, core features, and computational performance of this tool are demonstrated with clear and concise examples pertaining to different classes of molecular systems. We anticipate that PySAGES will provide the scientific community with a robust and easily accessible platform to accelerate simulations, improve sampling, and enable facile estimation of free energies for a wide range of materials and processes.
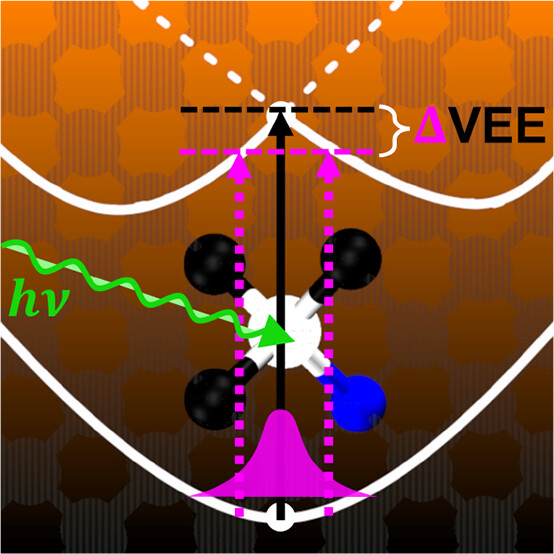
Quantum Vibronic Effects on the Excitation Energies of the Nitrogen-Vacancy Center in Diamond
Arpan Kundu and Giulia Galli
J. Phys. Chem. Lett., 15, 3, 802-810 (2024) DOI: 10.1021/acs.jpclett.3c03269
We investigated the impact of quantum vibronic coupling on the electronic properties of solid-state spin defects using stochastic methods and first-principles molecular dynamics with a quantum thermostat. Focusing on the negatively charged nitrogen-vacancy center in diamond as an exemplary case, we found a significant dynamic Jahn–Teller splitting of the doubly degenerate single-particle levels within the diamond’s band gap, even at 0 K, with a magnitude exceeding 180 meV. This pronounced splitting leads to substantial renormalizations of these levels and, subsequently, of the vertical excitation energies of the doubly degenerate singlet and triplet excited states. Our findings underscore the pressing need to incorporate quantum vibronic effects into first-principles calculations, particularly when comparing computed vertical excitation energies with experimental data. Our study also reveals the efficiency of stochastic thermal line sampling for studying phonon renormalizations of solid-state spin defects.
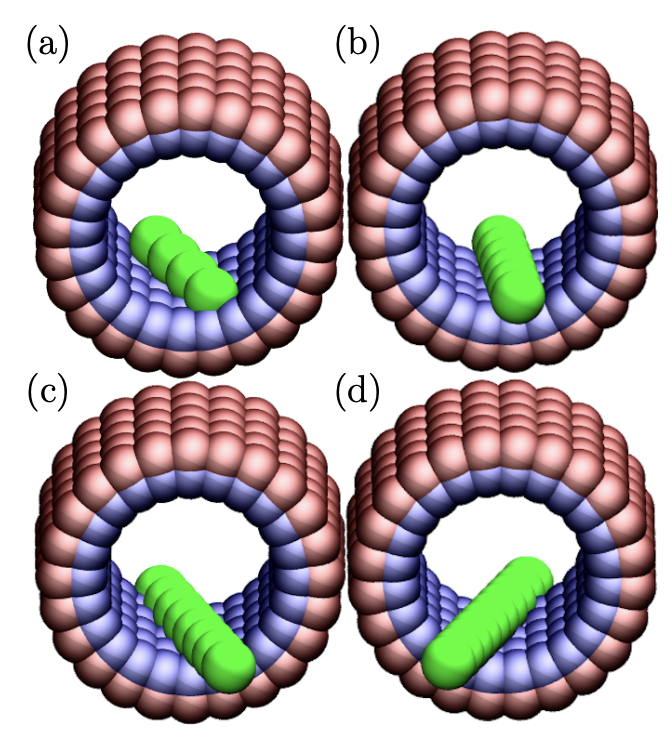
Calculating Binding Free Energies in Model Host-Guest Systems with Unrestrained Advanced Sampling
Andrew V. Marquardt, Mohsen Farshad, Jonathan K. Whitmer
J. Chem. Theory Comput. 20, 9, 3927-3934 (2024) DOI: 10.1021/acs.jctc.3c01186
Host--guest interactions are important to the design of pharmaceuticals, and more broadly to soft materials, as they can enable targeted, strong, and specific interactions between molecules. The binding process between host and guest may be classified as a ``rare event'' when viewing the system at atomic scales, such as those explored in molecular dynamics simulations. To obtain equilibrium binding conformations and dissociation constants from these simulations, it is essential to resolve such rare events. Advanced sampling methods such as Adaptive Biasing Force (ABF) promote the occurrence of less probable configurations in a system, therefore facilitating the sampling of essential collective variables (CVs) which characterize the host--guest interactions. Here, we present the application of ABF to a rod--cavitand coarse-grained (CG) model of host-guest systems to acquire the potential of mean force (PMF). We show that the employment of ABF enables the computation of configurational and thermodynamic properties of bound and unbound states, including the free energy landscape. Moreover, we identify important dynamical bottlenecks that limit sampling and discuss how these may be addressed in more general systems.
Raman Spectra of Electrified Si-Water Interfaces: First Principles Simulations
Zifan Ye, Francois Gygi, and Giulia Galli
J. Phys. Chem. Lett. 15. 1, 51-58 (2024) DOI: 10.1021/acs.jpclett.3c03122
We investigate the Raman spectra of liquid water in contact with a semiconductor surface using first-principles molecular dynamics simulations. We focus on a hydrogenated silicon–water interface and compute the Raman spectra from time correlation functions of the polarizability. We establish a relationship between Raman spectral signatures and structural properties of the liquid at the interface, and we identify the vibrational impacts of an applied electric field. We show that negative bias leads to a reduction of the number of hydrogen bonds (HBs) formed between the surface and the topmost water layer and an enhancement of the HB interactions between water molecules. Instead, positive bias leads to an enhancement of both the HB interactions between water and the surface and between water molecules, creating a semi-ordered interfacial layer. Our work provides molecular-level insights into electrified semiconductor/water interfaces and the identification of specific structural features through Raman spectroscopy.
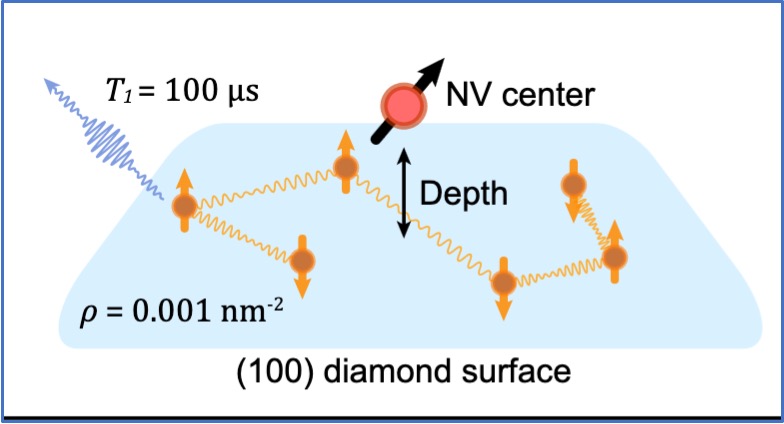
Understanding central spin decoherence due to interacting dissipative spin baths
Mykyta Onizhuk, Yu-Xin Wang, Jonah Nagura, Aashish A. Clerk, Giulia Galli
Phys. Rev. Lett., 132, 250401 (2024) DOI: 10.1103/PhysRevLett.132.250401
We propose a new approach to simulate the decoherence of a central spin coupled to an interacting dissipative spin bath with cluster-correlation expansion techniques. We benchmark the approach on generic 1D and 2D spin baths and find excellent agreement with numerically exact simulations. Our calculations show a complex interplay between dissipation and coherent spin exchange, leading to increased central spin coherence in the presence of fast dissipation. Finally, we model near-surface nitrogen-vacancy centers in diamond and show that accounting for bath dissipation is crucial to understanding their decoherence. Our method can be applied to a variety of systems and provides a powerful tool to investigate spin dynamics in dissipative environments.

Permutationally Invariant Networks for Enhanced Sampling (PINES): Discovery of Multimolecular and Solvent-Inclusive Collective Variables
Nicholas S. M. Herringer, Siva Dasetty, Diya Gandhi, Junhee Lee, Andrew L. Ferguson*
J. Chem. Theory Comput. 20, 1, 178-198 (2023). DOI: 10.1021/acs.jctc.3c00923
The typically rugged nature of molecular free-energy landscapes can frustrate efficient sampling of the thermodynamically relevant phase space due to the presence of high free-energy barriers. Enhanced sampling techniques can improve phase space exploration by accelerating sampling along particular collective variables (CVs). A number of techniques exist for the data-driven discovery of CVs parametrizing the important large-scale motions of the system. A challenge to CV discovery is learning CVs invariant to the symmetries of the molecular system, frequently rigid translation, rigid rotation, and permutational relabeling of identical particles. Of these, permutational invariance has proved a persistent challenge in frustrating the data-driven discovery of multimolecular CVs in systems of self-assembling particles and solvent-inclusive CVs for solvated systems. In this work, we integrate permutation invariant vector (PIV) featurizations with autoencoding neural networks to learn nonlinear CVs invariant to translation, rotation, and permutation and perform interleaved rounds of CV discovery and enhanced sampling to iteratively expand the sampling of configurational phase space and obtain converged CVs and free-energy landscapes. We demonstrate the permutationally invariant network for enhanced sampling (PINES) approach in applications to the self-assembly of a 13-atom argon cluster, association/dissociation of a NaCl ion pair in water, and hydrophobic collapse of a C45H92 n-pentatetracontane polymer chain. We make the approach freely available as a new module within the PLUMED2 enhanced sampling libraries.
Excited state properties of point defects in semiconductors and insulators investigated with time-dependent density functional theory
Yu Jin, Victor Wen-zhe Yu, Marco Govoni, Andrew C. Xu, and Giulia Galli
J. Chem. Theory Comput. 19, 8689–8705 (2023). DOI: 10.1021/acs.jctc.3c00986
We present a formulation of spin-conserving and spin-flip hybrid time-dependent density functional theory (TDDFT), including the calculation of analytical forces, which allows for efficient calculations of excited state properties of solid-state systems with hundreds to thousands of atoms. We discuss an implementation on both GPU- and CPU-based architectures along with several acceleration techniques. We then apply our formulation to the study of several point defects in semiconductors and insulators, specifically the negatively charged nitrogen-vacancy and neutral silicon-vacancy centers in diamond, the neutral divacancy center in 4H silicon carbide, and the neutral oxygen-vacancy center in magnesium oxide. Our results highlight the importance of taking into account structural relaxations in excited states in order to interpret and predict optical absorption and emission mechanisms in spin defects.
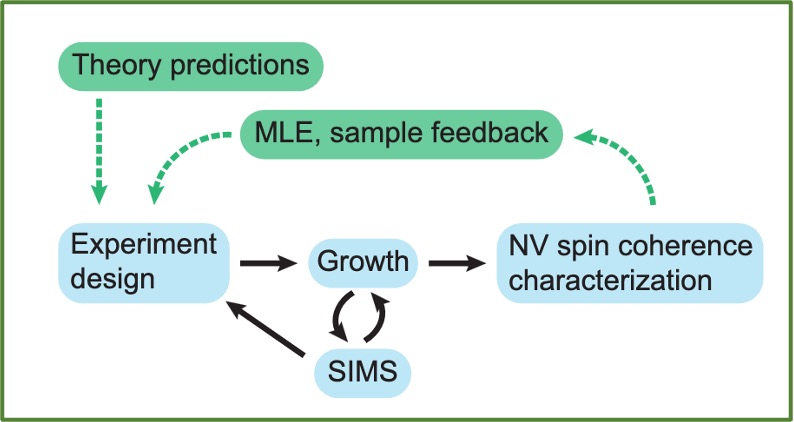
Guiding Diamond Spin Qubit Growth with Computational Methods
Jonathan C. Marcks, Mykyta Onizhuk, Nazar Delegan, Yu-Xin Wang, Masaya Fukami, Maya Watts, Aashish A. Clerk, F. Joseph Heremans, Giulia Galli, and David D. Awschalom
Phys. Rev. Materials 8, 026204 (2024). DOI:10.1103/PhysRevMaterials.8.026204
The nitrogen vacancy (NV) center in diamond, a well-studied, optically active spin defect, is the prototypical system in many state of the art quantum sensing and communication applications. In addition to the enticing properties intrinsic to the NV center, its diamond host's nuclear and electronic spin baths can be leveraged as resources for quantum information, rather than considered solely as sources of decoherence. However, current synthesis approaches result in stochastic defect spin positions, reducing the technology's potential for deterministic control and yield of NV-spin bath systems, as well as scalability and integration with other technologies. Here, we demonstrate the use of theoretical calculations of electronic central spin decoherence as an integral part of an NV-spin bath synthesis workflow, providing a path forward for the quantitative design of NV center-based quantum sensing systems. We use computationally generated coherence data to characterize the properties of single NV center qubits across relevant growth parameters to find general trends in coherence time distributions dependent on spin bath dimensionality and density. We then build a maximum likelihood estimator with our theoretical model, enabling the characterization of a test sample through NV T2* measurements. Finally, we explore the impact of dimensionality on the yield of strongly coupled electron spin systems. The methods presented herein are general and applicable to other qubit platforms that can be appropriately simulated.
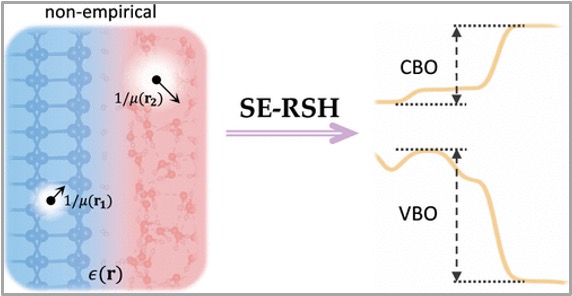
Nonempirical Range-Separated Hybrid Functional with Spatially Dependent Screened Exchange
Jiawei Zhan, Marco Govoni, and Giulia Galli
J. Chem. Theory Comput. 19, 5851-5862 (2023). DOI: 10.1038/s41467-023-41632-9
Electronic structure calculations based on density functional theory (DFT) have successfully predicted numerous ground-state properties of a variety of molecules and materials. However, exchange and correlation functionals currently used in the literature, including semilocal and hybrid functionals, are often inaccurate to describe the electronic properties of heterogeneous solids, especially systems composed of building blocks with large dielectric mismatch. Here, we present a dielectric-dependent range-separated hybrid functional, screened-exchange range-separated hybrid (SE-RSH), for the investigation of heterogeneous materials. We define a spatially dependent fraction of exact exchange inspired by the static Coulomb-hole and screened-exchange (COHSEX) approximation used in many-body perturbation theory, and we show that the proposed functional accurately predicts the electronic structure of several nonmetallic interfaces, three- and two-dimensional, pristine, and defective solids and nanoparticles.
Engineering the formation of spin-defects from first principles
Cunzhi Zhang, Francois Gygi & Giulia Galli
Nat. Commun. 14, 5985 (2023). DOI: 10.1038/s41467-023-41632-9
The full realization of spin qubits for quantum technologies relies on the ability to control and design the formation processes of spin defects in semiconductors and insulators. We present a computational protocol to investigate the synthesis of point-defects at the atomistic level, and we apply it to the study of a promising spin-qubit in silicon carbide, the divacancy (VV). Our strategy combines electronic structure calculations based on density functional theory and enhanced sampling techniques coupled with first principles molecular dynamics. We predict the optimal annealing temperatures for the formation of VVs at high temperature and show how to engineer the Fermi level of the material to optimize the defect’s yield for several polytypes of silicon carbide. Our results are in excellent agreement with available experimental data and provide novel atomistic insights into point defect formation and annihilation processes as a function of temperature.
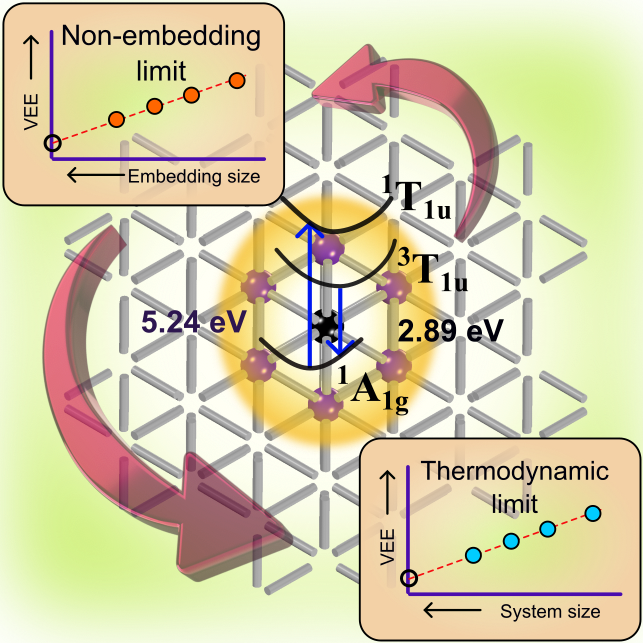
Optical Properties of Neutral F Centers in Bulk MgO with Density Matrix Embedding
Shreya Verma, Abhishek Mitra, Yu Jin, Soumi Haldar, Christian Vorwerk, Matthew R. Hermes, Giulia Galli, Laura Gagliardi
J. Phys. Chem. Lett., 14, 34, 7703-7710 (2023). DOI: 10.1021/acs.jpclett.3c01875
The optical spectra of neutral oxygen vacancies (F0 centers) in the bulk MgO lattice was investigated using density matrix embedding theory. The impurity Hamiltonian was solved with the complete active space self-consistent field (CAS-DMET) and second-order n-electron valence state perturbation theory (NEVPT2-DMET) multireference methods. To estimate defect-localized vertical excitation energies at the non-embedding and thermodynamic limits, a double extrapolation scheme was employed. The extrapolated NEVPT2-DMET vertical excitation energy value of 5.24 eV agrees well with the experimental absorption maxima at 5.03 eV, whereas the excitation energy value of 2.89 eV at the relaxed triplet defect localized state geometry overestimates the experimental emission at 2.4 eV by only nearly 0.5 eV, indicating the involvement of triplet-singlet decay pathway.
Quantum vibronic effects on the electronic properties of molecular crystals
Arpan Kundu and Giulia Galli
J. Chem. Theory Comput., 19, 13, 4011-4022 (2023). DOI: 10.1021/acs.jctc.3c00424
We present a study of molecular crystals, focused on the effect of nuclear quantum motion and anharmonicity on their electronic properties. We consider a system composed of relatively rigid molecules, a diamondoid crystal, and one composed of floppier molecules, NAI-DMAC, a thermally activated delayed fluorescence compound. We compute fundamental electronic gaps at the DFT level of theory, with the PBE and SCAN functionals, by coupling first-principles molecular dynamics with a nuclear quantum thermostat. We find a sizable zero-point-renormalization (ZPR) of the band gaps, which is much larger in the case of diamondoids ( 0.6 eV) than for NAI-DMAC ( 0.22 eV). We show that the frozen phonon (FP) approximation, which neglects intermolecular anharmonic effects, leads to a large error (∼ 50%) in the calculation of the band gap ZPR. Instead, when using a stochastic method, we obtain results in good agreement with those of our quantum simulations for the diamondoid crystal. However, the agreement is worse for NAI-DMAC where intra-molecular anharmonicities contribute to the ZPR. Our results highlight the importance of accurately including nuclear and anharmonic quantum effects to predict the electronic properties of molecular crystals.
Thermal Conductivity of Water at Extreme Conditions
Cunzhi Zhang, Marcello Puligheddu, Linfeng Zhang, Roberto Car, Giulia Galli
J. Phys. Chem. B 127, 31, 7011-7017 (2023). DOI: 10.1021/acs.jpcb.3c02972
Measuring the thermal conductivity (κ) of water at extreme conditions is a challenging task and few experimental data are available. We predict κ for temperatures and pressures relevant to the conditions of the Earth mantle, between 1,000 and 2,000 K and up to 22 GPa. We employ close to equilibrium molecular dynamics simulations and a deep neural network potential fitted to density functional theory data. We then interpret our results by computing the equation of state of water on a fine grid of points and using a simple model for κ. We find that the thermal conductivity is weakly dependent on temperature and monotonically increases with pressure with an approximate square-root behavior. In addition we show how the increase of κ at high pressure, relative to ambient conditions, is related to the corresponding increase in the sound velocity. Although the relationships between the thermal conductivity, pressure and sound velocity established here are not rigorous, they are sufficiently accurate to allow for a robust estimate of the thermal conductivity of water in a broad range of temperatures and pressures, where experiments are still difficult to perform.
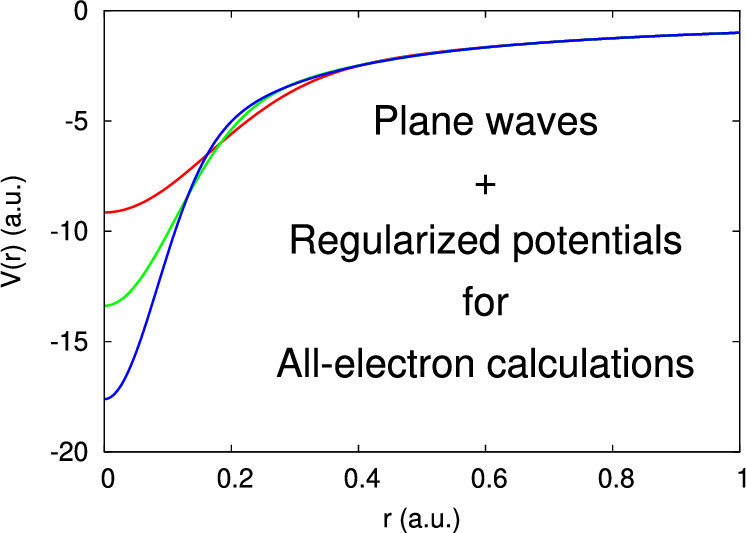
All-Electron Plane-Wave Electronic Structure Calculations
Francois Gygi
J. Chem. Theory Comput. 19, 4, 1300-1309 (2023). DOI: 10.1021/acs.jctc.2c01191
We demonstrate the use of the plane wave basis for all-electron electronic structure calculations. The approach relies on the definition of an analytic, norm-conserving, regularized Coulomb potential, and a scalable implementation of the plane wave method capable of handling large energy cutoffs (up to 80 kRy in the examples shown). The method is applied to the computation of electronic properties of isolated atoms as well as the diamond and silicon crystals, MgO, solid argon, and a configuration of 64 water molecules extracted from a first-principles molecular dynamics simulation. The computed energies, band gaps, ionic forces, and stress tensors provide reference results for the validation of pseudopotentials and/or localized basis sets. A calculation of the all-electron band structure of diamond and silicon using the SCAN meta-GGA density functional allows for a validation of calculations based on pseudopotentials derived using the PBE exchange-correlation functional. In the case of (H2O)64, the computed ionic forces provide a reference from which the errors incurred in pseudopotential calculations and in localized Gaussian basis sets calculations can be estimated.
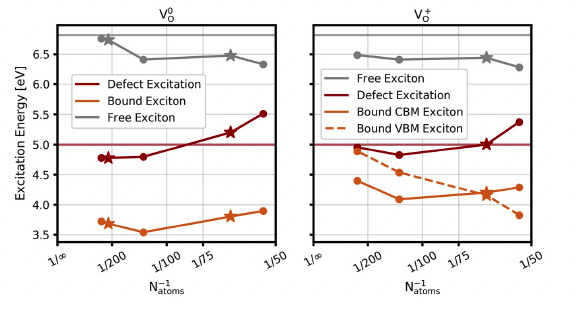
Disentangling photoexcitation and photoluminescence processes in defective MgO
Christian Vorverk and Giulia Galli
Phys. Rev. Mat., 7, 033801 (2023). DOI: 10.1103/PhysRevMaterials.7.033801
Oxygen vacancies are ubiquitous in oxides and strongly influence the material's electronic structure and catalytic and transport properties. Here we focus on a seemingly simple defective oxide, MgO, and on the electronic properties of oxygen vacancies, which remain controversial in spite of numerous studies. We present an ab initio investigation of the photoexcitation and photoionization process of these defects, using a newly developed embedding Bethe-Salpeter equation approach, implemented in the WEST code. We find absorption and emission energies in good agreement with experiments. Our results provide a detailed, microscopic understanding of the absorption and emission processes of the neutral and positively charged oxygen vacancy, reconciling different views present in the chemistry and condensed-matter physics communities.
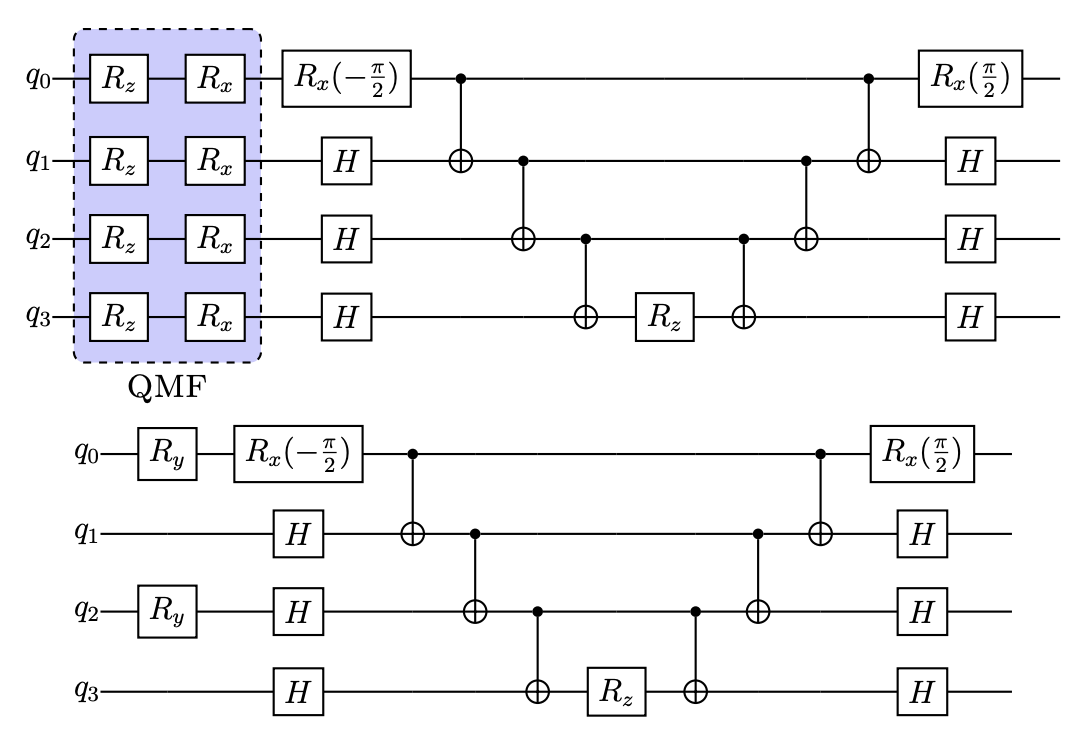
Quantum simulations of Fermionic Hamiltonians with efficient encoding and ansatz schemes
Benchen Huang, Nan Sheng, Marco Govoni, and Giulia Galli
J. Chem. Theory Comput., 19, 5, 1487–1498 (2023). DOI: 10.1021/acs.jctc.2c01119
We propose a computational protocol for quantum simulations of Fermionic Hamiltonians on a quantum computer, enabling calculations which were previously not feasible with conventional encoding and ansatses of variational quantum eigensolvers (VQE). We combine a qubit-efficient encoding scheme mapping Slater determinants onto qubits with a modified qubit-coupled cluster ansatz and noise-mitigation techniques. Our strategy leads to a substantial improvement in the scaling of circuit gate counts and to a decrease in the number of required variational parameters, thus increasing the resilience to noise. We present results for spin defects of interest for quantum technologies, going beyond minimum models for the negatively charged nitrogen vacancy center in diamond and the double vacancy in 4H silicon carbide (4H-SiC) and tackling a defect as complex as negatively charged silicon vacancy in 4H-SiC for the first time.
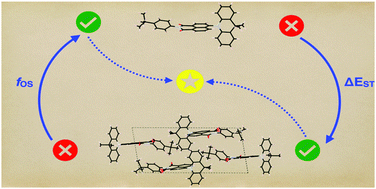
Quantum simulations of thermally activated delayed fluorescence in an all-organic emitter
Tommaso Francese, Arpan Kundu, Francois Gygi and Giulia Galli
Phys. Chem. Chem. Phys., 24, 10101-10113 (2022). DOI:10.1039/D2CP01147F
We investigate the prototypical NAI-DMAC thermally activated delayed fluorescence (TADF) emitter in the gas phase- and high-packing fraction limits at finite temperature, by combining first principles molecular dynamics with a quantum thermostat to account for nuclear quantum effects (NQE). We find a weak dependence of the singlet–triplet energy gap (ΔEST) on temperature in both the solid and the molecule, and a substantial effect of packing. While the ΔEST vanishes in the perfect crystal, it is of the order of ∼0.3 eV in the molecule, with fluctuations ranging from 0.1 to 0.4 eV at 300 K. The transition probability between the HOMOs and LUMOs has a stronger dependence on temperature than the singlet–triplet gap, with a desirable effect for thermally activated fluorescence; such temperature effect is weaker in the condensed phase than in the molecule. Our results on ΔEST and oscillator strengths, together with our estimates of direct and reverse intersystem crossing rates, show that optimization of packing and geometrical conformation is critical to increase the efficiency of TADF compounds. Our findings highlight the importance of considering thermal fluctuations and NQE to obtain robust predictions of the electronic properties of NAI-DMAC.
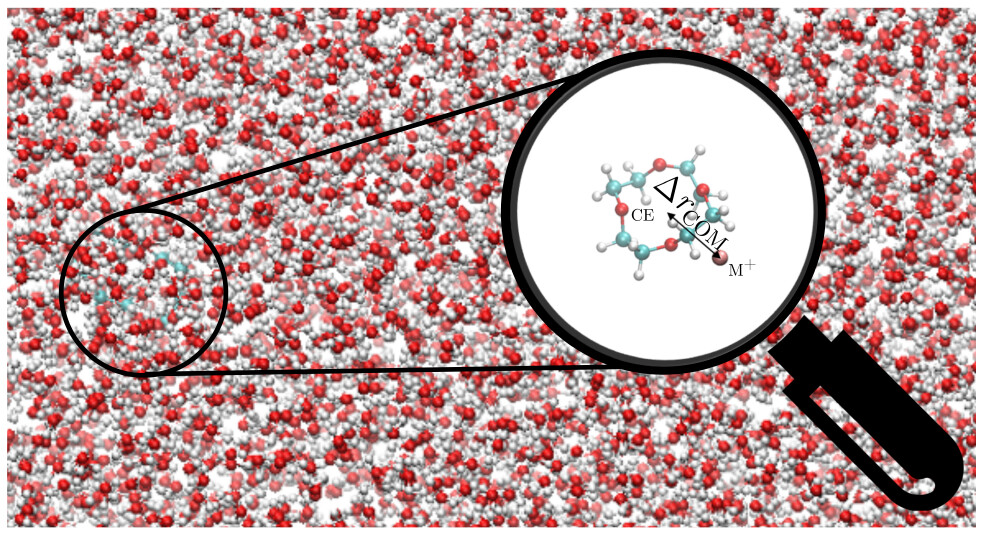
Thermodynamics of Li+–Crown Ether Interactions in Aqueous Solvent
Ramón González-Pérez, Stephen Adams, Alexander W. Dowling, William A. Phillip, Jonathan Whitmer
J. Phys. Chem. A 127, 21, 4624-4631 (2023). DOI:10.1021/acs.jpca.3c00094
Lithium ion-based batteries are ubiquitous in modern technology due to applications in personal electronics and high-capacity storage for electric vehicles. Concerns about lithium supply and battery waste have prompted interest in lithium recycling methods. The crown ether, 12-crown-4, has been studied for its abilities to form stable complexes with lithium ions (\ce{Li+}). In this paper, molecular dynamics simulations are applied to examine the binding properties of a 12-crown-4—Li+ system in aqueous solution. It was found that 12-crown-4 did not form stable complexes with Li+ in aqueous solution due to the binding geometry which was prone to interference by surrounding water molecules. In addition, the binding properties of sodium ions (Na+) to 12-crown-4 are examined for comparison. Subsequently, calculations were performed with the crown ethers 15-crown-5 and 18-crown-6 to study their complexation with Li+ as well. It was determined that binding was unfavorable for both types of ions for all three crown ethers tested, though 15-crown-5 and 18-crown-6 showed a marginally greater affinity for Li+ than 12-crown-4. Metastable minima present in the potential of mean force for Na+ render binding marginally more likely there. We discuss these results in the context of membrane based applications of crown ethers for Li+ separations.
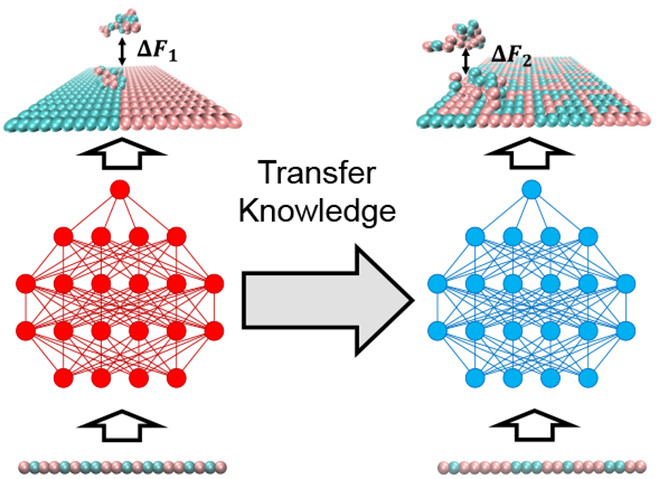
Transfer Learning Facilitates the Prediction of Polymer-Surface Adhesion Strength
Jiale Shi, Fahed Albreiki, Yamil J. Colón, Samanvaya Srivastava, Jonathan K. Whitmer
J. Chem. Theory Comput., 19, 14, 4631-4640 (2023). DOI:10.1021/acs.jctc.2c01314
Machine learning (ML) accelerates the exploration of material properties and their links to the structure of the underlying molecules. In previous work [J. Shi, M. J. Quevillon, P. H. A. Valença, and J. K. Whitmer, \textit{ACS Appl. Mater. Interfaces.}, 2022, 14, 32, 37161--37169], ML models were applied to predict the adhesive free energy of polymer--surface interactions with high accuracy from the knowledge of the sequence data, demonstrating successes in inverse-design of polymer sequence for known surface compositions. While the method was shown to be successful in designing polymers for a known surface, extensive datasets were needed for each specific surface in order to train the surrogate models. Ideally, one should be able to infer information about similar surfaces without having to regenerate a full complement of adhesion data for each new case. In the current work, we demonstrate a transfer learning (TL) technique using a deep neural network to improve the accuracy of ML models trained on small datasets by pre-training on a larger database from a related system and fine-tuning the weights of all layers with a small amount of additional data. The shared knowledge from the pre-trained model facilitates the prediction accuracy significantly on small datasets. We also explore the limits of database size on accuracy and the optimal tuning of network architecture and parameters for our learning tasks. While applied to a relatively simple coarse-grained (CG) polymer model, the general lessons of this study apply to detailed modeling studies and the broader problems of inverse materials design.
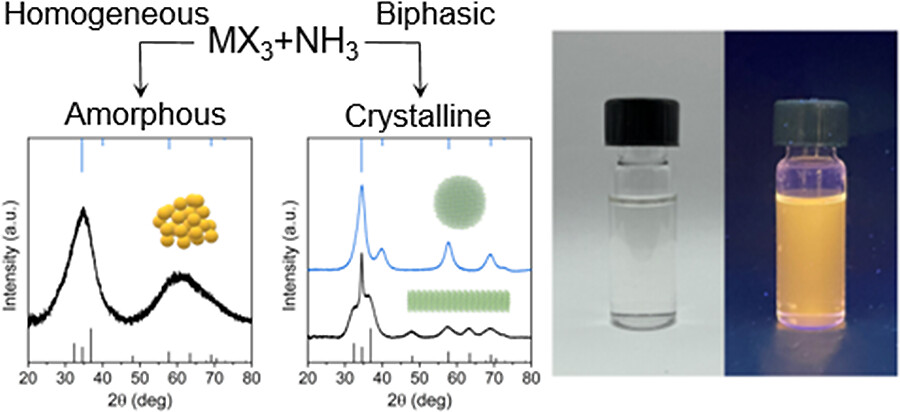
Synthesis of Colloidal GaN and AlN Nanocrystals in Biphasic Molten Salt/Organic Solvent Mixtures under High-Pressure Ammonia
Wooje Cho, Zirui Zhou, Ruiming Lin, Justin C. Ondry, and Dmitri V. Talapin
ACS Nano 17, 2, 1315–1326 (2023). DOI:10.1021/acsnano.2c09552
Group III nitrides are of great technological importance for electronic devices. These materials have been widely manufactured via high-temperature methods such as physical vapor transport (PVT), chemical vapor deposition (CVD), and hydride vapor phase epitaxy (HVPE). The preparation of group III nitrides by colloidal synthesis methods would provide significant advantages in the form of optical tunability via size and shape control and enable cost reductions through scalable solution-based device integration. Solution syntheses of III-nitride nanocrystals, however, have been scarce, and the quality of the synthesized products has been unsatisfactory for practical use. Here, we report that incorporating a molten salt phase in solution synthesis can provide a viable option for producing crystalline III-nitride nanomaterials. Crystalline GaN and AlN nanomaterials can be grown in a biphasic molten-salt/organic-solvent mixture under an ammonia atmosphere at moderate temperatures (less than 300 °C) and stabilized under ambient conditions by postsynthetic treatment with organic surface ligands. We suggest that microscopic reversibility of monomer attachment, which is essential for crystalline growth, can be achieved in molten salt during the nucleation and the growth of the III-nitride nanocrystals. We also show that increased ammonia pressure increases the size of the GaN nanocrystals produced. This work demonstrates that use of molten salt and high-pressure reactants significantly expands the chemical scope of solution synthesis of inorganic nanomaterials.
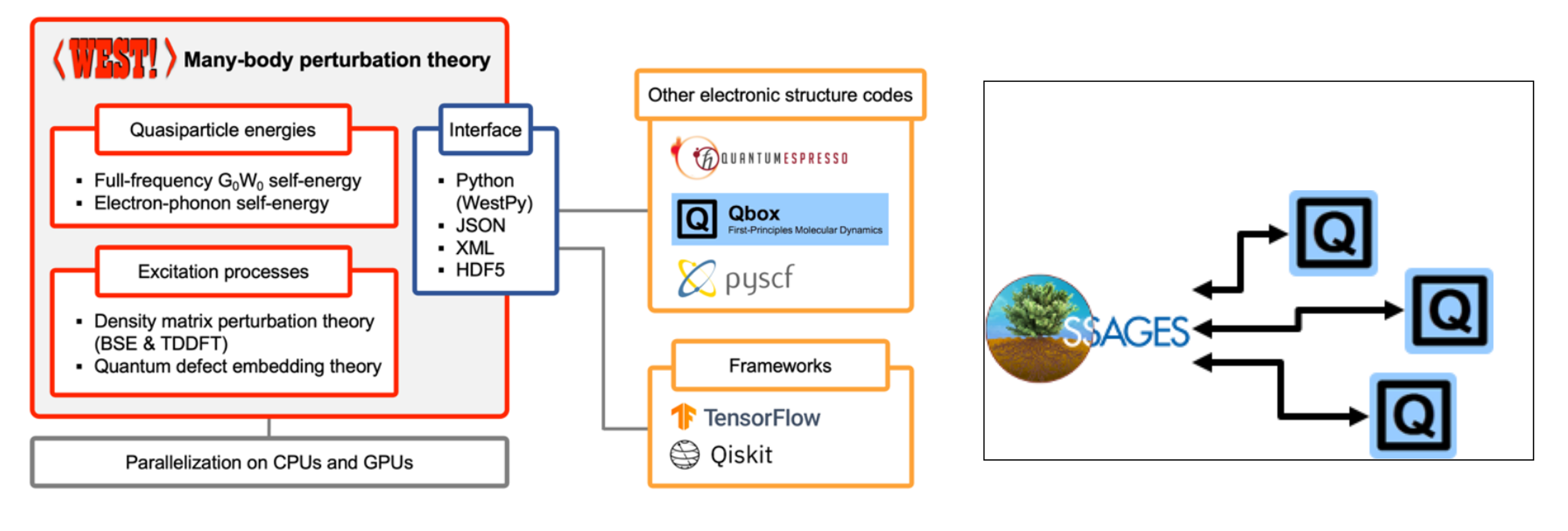
Roadmap on Electronic Structure Codes in the Exascale Era
Vikram Gavini, [...], Giulia Galli, [...], Marco Govoni, [...], Francois Gygi, [...], and Danny Perez
Modelling Simul. Mater. Sci. Eng. 31, 063301 (2022). DOI: 10.1088/1361-651X/acdf06
Electronic structure calculations have been instrumental in providing many important insights into a range of physical and chemical properties of various molecular and solid-state systems. Their importance to various fields, including materials science, chemical sciences, computational chemistry and device physics, is underscored by the large fraction of available public supercomputing resources devoted to these calculations. As we enter the exascale era, exciting new opportunities to increase simulation numbers, sizes, and accuracies present themselves. In order to realize these promises, the community of electronic structure software developers will however first have to tackle a number of challenges pertaining to the efficient use of new architectures that will rely heavily on massive parallelism and hardware accelerators. This roadmap provides a broad overview of the state-of-the-art in electronic structure calculations and of the various new directions being pursued by the community. It covers 14 electronic structure codes, presenting their current status, their development priorities over the next five years, and their plans towards tackling the challenges and leveraging the opportunities presented by the advent of exascale computing.
Current Rectification by Nanoparticles in Bipolar Nanopores
Andres Cordoba, Joan M. de Oca, Johnson Dhanasekaran, Seth B. Darling, and Juan J. de Pablo
Mol. Syst. Des. Eng. 8, 289-299 (2023). DOI:10.1039/D2ME00187J
Bipolar nanochannels comprising two domains of positively and negatively charged walls along the pore axis are known to rectify current when exposed to an electric potential bias. We find that addition of charged nanoparticles can increase rectification considerably, by approximately one order of magnitude. Two bipolar channel geometries are considered here; their behavior is examined at rest and under the influence of a negative bias and a positive bias, respectively. We do so by relying on a molecular-level model of the electrolyte solution in the channels. The large increase in current rectification can be explained by the inherent electric field that charged nanoparticles generate within the channel. This effect is found to be largely dependent on the pore's geometry, its charge distribution, and the sign of the nanoparticles' charge, thereby offering new opportunities for design of engineered nanopore membrane-nanoparticle systems for energy storage.
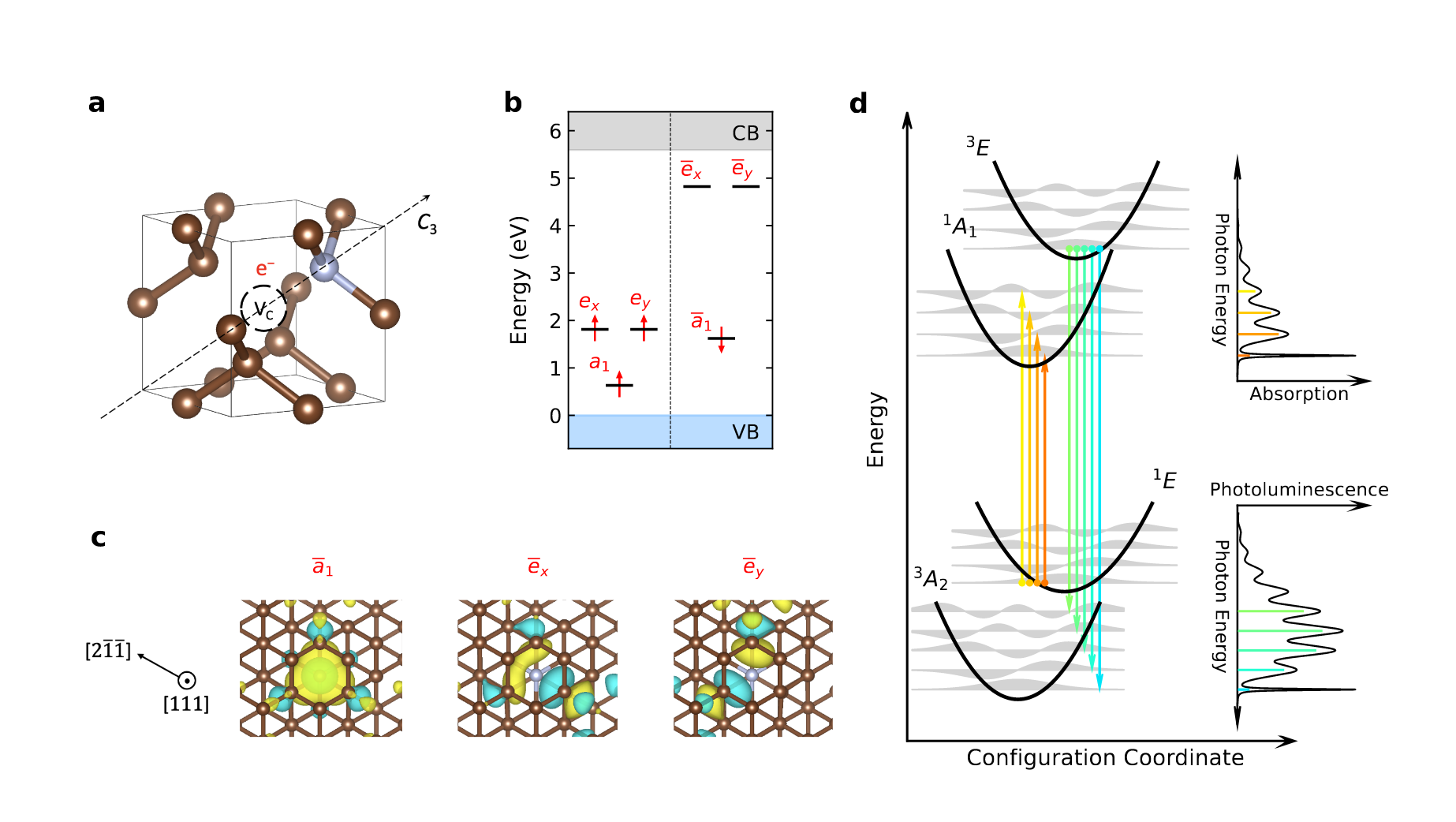
Vibrationally resolved optical excitations of the nitrogen-vacancy center in diamond
Yu Jin, Marco Govoni, and Giulia Galli
npj Comput. Mater. 8, 238 (2022). DOI:10.1038/s41524-022-00928-y
A comprehensive description of the optical cycle of spin defects in solids requires the understanding of the electronic and atomistic structure of states with different spin multiplicity, including singlet states which are particularly challenging from a theoretical standpoint. We present a general framework, based on spin-flip time-dependent density function theory, to determine the excited state potential energy surfaces of the many-body singlet states of spin defects; we then predict the vibrationally resolved absorption spectrum between singlet shelving states of a prototypical defect, the nitrogen-vacancy center in diamond. Our results, which are in very good agreement with experiments, provide an interpretation of the measured spectra and reveal the key role of specific phonons in determining absorption processes, and the notable influence of non-adiabatic interactions. The insights gained from our calculations may be useful in defining strategies to improve infrared-absorption-based magnetometry and optical pumping schemes. The theoretical framework developed here is general and applicable to a variety of other spin defects and materials.
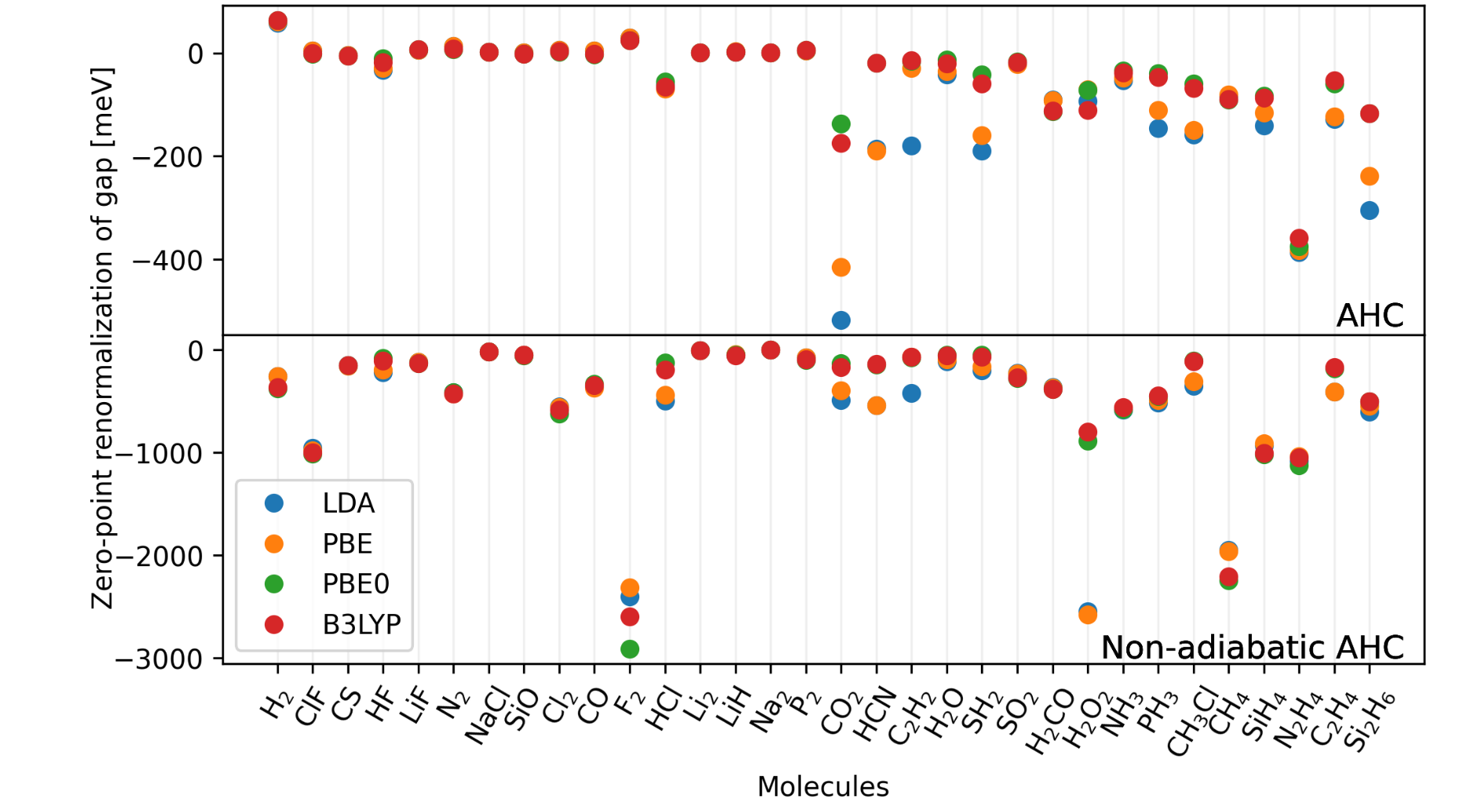
Computational Protocol to Evaluate Electron-Phonon Interactions within Density Matrix Perturbation Theory
Han Yang, Marco Govoni, Arpan Kundu, and Giulia Galli
J. Chem. Theory Comput. 18, 10, 6031 (2022). DOI:10.1021/acs.jctc.2c00579
We present a computational protocol, based on density matrix perturbation theory, to obtain non-adiabatic, frequency-dependent electron–phonon self-energies for molecules and solids. Our approach enables the evaluation of electron–phonon interaction using hybrid functionals, for spin-polarized systems, and the computational overhead to include dynamical and non-adiabatic terms in the evaluation of electron–phonon self-energies is negligible. We discuss results for molecules, as well as pristine and defective solids.
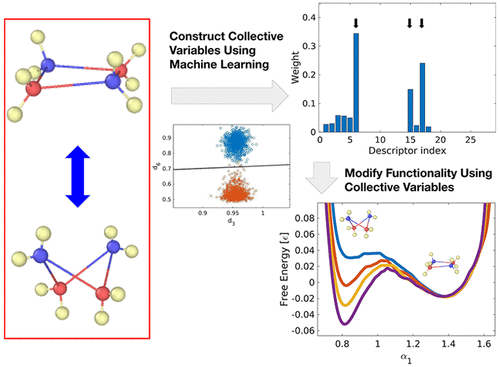
Collective Variables for Free Energy Surface Tailoring: Understanding and Modifying Functionality in Systems Dominated by Rare Events
Dan Mendels and Juan J. de Pablo
J. Phys. Chem. Lett. 13, 12, 2830-2837 (2022). DOI:10.1021/acs.jpclett.2c00317
We introduce a method for elucidating and modifying the functionality of systems dominated by rare events that relies on the semiautomated tuning of their underlying free energy surface. The proposed approach seeks to construct collective variables (CVs) that encode the essential information regarding the rare events of the system of interest. The appropriate CVs are identified using harmonic linear discriminant analysis (HLDA), a machine-learning-based method that is trained solely on data collected from short ordinary simulations in the relevant metastable states of the system. Utilizing the interpretable form of the resulting CVs, the critical interaction potentials that determine the system’s rare transitions are identified and purposely modified to tailor the free energy surface in a manner that alters functionality as desired. The applicability of the method is illustrated in the context of three different systems, thereby demonstrating that thermodynamic and kinetic properties can be tractably modified with little to no prior knowledge or intuition.
Ionic Transport in Electrostatic Janus Membranes. An Explicit Solvent Molecular Dynamic Simulation
Joan M. Montes de Oca, Johnson Dhanasekaran, Andres Cordoba, Seth B. Darling, and Juan J. de Pablo
ACS Nano 16, 3, 3768 (2022). DOI:10.1021/acsnano.1c07706
Janus, or two-sided, charged membranes offer promise as ionic current rectifiers. In such systems, pores consisting of two regions of opposite charge can be used to generate a current from a gradient in salinity. The efficiency of nanoscale Janus pores increases dramatically as their diameter becomes smaller. However, little is known about the underlying transport processes, particularly under experimentally accessible conditions. In this work, we examine the molecular basis for rectification in Janus nanopores using an applied electric field. Molecular simulations with explicit water and ions are used to examine the structure and dynamics of all molecular species in aqueous electrolyte solutions. For several macroscopic observables, the results of such simulations are consistent with experimental observations on asymmetric membranes. Our analysis reveals a number of previously unknown features, including a pronounced local reorientation of water molecules in the pores, and a segregation of ionic species that had not been anticipated by previously reported continuum analyses of Janus pores. Using these insights, a model is proposed for ionic current rectification in which electric leakage at the pore entrance controls net transport.
Influence of Nuclear Quantum Effects on the Electronic Properties of Amorphous Carbon
Arpan Kundu, Yunxiang Song, and Giulia Galli
PNAS 119 (31), e2203083119 (2022). DOI: 10.1073/pnas.220308311
We carry out quantum simulations to study the physical properties of diamond-like amorphous carbon by coupling first-principles molecular dynamics with a quantum thermostat, and we analyze multiple samples representative of different defective sites present in the disordered network. We show that quantum vibronic coupling is critical in determining the electronic properties of the system, in particular its electronic and mobility gaps, while it has a moderate influence on the structural properties. We find that despite localized electronic states near the Fermi level, the quantum nature of the nuclear motion leads to a renormalization of the electronic gap surprisingly similar to that found in crystalline diamond. We also discuss the notable influence of nuclear quantum effects on band-like and variable-hopping mechanisms contributing to electrical conduction. Our calculations indicate that methods often used to evaluate electron–phonon coupling in ordered solids are inaccurate to study the electronic and transport properties of amorphous semiconductors composed of light atoms.
Systematic Modification of Functionality in Disordered Elastic Networks Through Free Energy Surface Tailoring
Dan Mendels, Fabian Bylehn, Timothy W. Sirk, and Juan J. de Pablo
Science Advances 9, 23 (2023). DOI:10.1126/sciadv.adf7541
A combined machine learning–physics–based approach is explored for molecular and materials engineering. Specifically, collective variables, akin to those used in enhanced sampled simulations, are constructed using a machine learning model trained on data gathered from a single system. Through the constructed collective variables, it becomes possible to identify critical molecular interactions in the considered system, the modulation of which enables a systematic tailoring of the system’s free energy landscape. To explore the efficacy of the proposed approach, we use it to engineer allosteric regulation and uniaxial strain fluctuations in a complex disordered elastic network. Its successful application in these two cases provides insights regarding how functionality is governed in systems characterized by extensive connectivity and points to its potential for design of complex molecular systems.
DFT Exchange: Sharing Perspectives on the Workhorse of Quantum Chemistry and Materials Science
Andrew Teale, [...], Giulia Galli, [...], Weitao Yang
Phys. Chem. Chem. Phys. 24, 28700-28781 (2022). DOI: 10.1039/D2CP02827A
In this paper, the history, present status, and future of density-functional theory (DFT) is informally reviewed and discussed by 70 workers in the field, including molecular scientists, materials scientists, method developers and practitioners. The format of the paper is that of a roundtable discussion, in which the participants express and exchange views on DFT in the form of 302 individual contributions, formulated as responses to a preset list of 26 questions. Supported by a bibliography of 777 entries, the paper represents a broad snapshot of DFT, anno 2022.
Diffusion-Limited Kinetics of Isovalent Cation Exchange in III-V Nanocrystals Dispersed in Molten Salt Reaction Media.
A. Gupta, J. Ondry, M. Chen, M. Hudson, I. Coropceanu, N. Sarma, D.V. Talapin
Nano Lett. 22, 16, 6545 (2022). DOI:10.1021/acs.nanolett.2c01699
Ternary III-V quantum dots can be synthesized by cation exchange in binary III-V colloidal quantum dots using molten salts as the solvent and cation source. The goal of this work is to determine the kinetic factors which govern isovalent cation exchange. We focus on the reactions of InP+GaI3 à In1-xGaxP and InAs+GaI3 à In1-xGaxAs to create technologically important ternary III-V phases. We find that the molten salt reaction medium causes the transformation of sphere-shaped InP nanocrystals to tetrahedron-shaped In1-xGaxP nanocrystals. Further, we determine that the activation energy for the cation exchange reaction is 0.9 eV for incorporation of Ga into InP and 1.17 eV for incorporation of Ga into InAs, both much smaller than the measured values in bulk semiconductors. Next, we use powder XRD simulations to constrain our understanding of the structure of the In1-xGaxP nanocrystals studied here. We find that both the initial InP nanocrystals and the resulting In1-xGaxP nanocrystals in this study contain considerable stacking disorder for the close pack which is unaffected by molten salt processing. Further we show that PXRD alone is insensitive to the structural parameters other than Ga-to-In ratio, such as potential core/shell morphologies or strain for the small crystallites studied here. Finally, we demonstrate that PXRD is insensitive to compositional variation among the individual In1-xGaxP nanocrystals in an ensemble, and then we develop a HRTEM based routine to elucidate the particle-to-particle variation of the composition.
Self-assembly of nanocrystals into strongly electronically coupled all-inorganic supercrystals
Igor Coropceanu, Eric M. Janke, Joshua Portner, Danny Haubold, Trung Dac Nguyen, Avishek Das, Christian P.N. Tanner, James K. Utterback, Samuel W. Teitelbaum, Margaret H. Hudson, Nivedina A. Sarma, Alex M. Hinkle, Christopher J. Tassone, Alexander Eychmüller, David T. Limmer, Monica Olvera De La Cruz, Naomi S. Ginsberg, and Dmitri V. Talapin
Science 375, 1422-1426 (2022). DOI: 10.1126/science.abm6753
Colloidal nanocrystals of metals, semiconductors, and other functional materials can self-assemble into long-range ordered crystalline and quasicrystalline phases, but insulating organic surface ligands prevent the development of collective electronic states in ordered nanocrystal assemblies. We reversibly self-assembled colloidal nanocrystals of gold, platinum, nickel, lead sulfide, and lead selenide with conductive inorganic ligands into supercrystals exhibiting optical and electronic properties consistent with strong electronic coupling between the constituent nanocrystals. The phase behavior of charge-stabilized nanocrystals can be rationalized and navigated with phase diagrams computed for particles interacting through short-range attractive potentials. By finely tuning interparticle interactions, the assembly was directed either through one-step nucleation or nonclassical two-step nucleation pathways. In the latter case, the nucleation was preceded by the formation of two metastable colloidal fluids.
GPU Acceleration of Large-Scale Full-Frequency GW Calculations
Victor Wen-zhe Yu and Marco Govoni
J. Chem. Theory Comput. 18, 8, 4690 (2022) DOI:10.1021/acs.jctc.2c00241
Many-body perturbation theory is a powerful method to simulate electronic excitations in molecules and materials starting from the output of density functional theory calculations. By implementing the theory efficiently so as to run at scale on the latest leadership high-performance computing systems it is possible to extend the scope of GW calculations. We present a GPU acceleration study of the full-frequency GW method as implemented in the WEST code. Excellent performance is achieved through the use of (i) optimized GPU libraries, e.g., cuFFT and cuBLAS, (ii) a hierarchical parallelization strategy that minimizes CPU-CPU, CPU-GPU, and GPU-GPU data transfer operations, (iii) asynchronous MPI communications that overlap with GPU computations, and (iv) mixed-precision in selected portions of the code. A series of performance benchmarks have been carried out on leadership high-performance computing systems, showing a substantial speedup of the GPU-accelerated version of WEST with respect to its CPU version. Good strong and weak scaling is demonstrated using up to 25920 GPUs. Finally, we showcase the capability of the GPU version of WEST for large-scale, full-frequency GW calculations of realistic systems, e.g., a nanostructure, an interface, and a defect, comprising up to 10368 valence electrons.
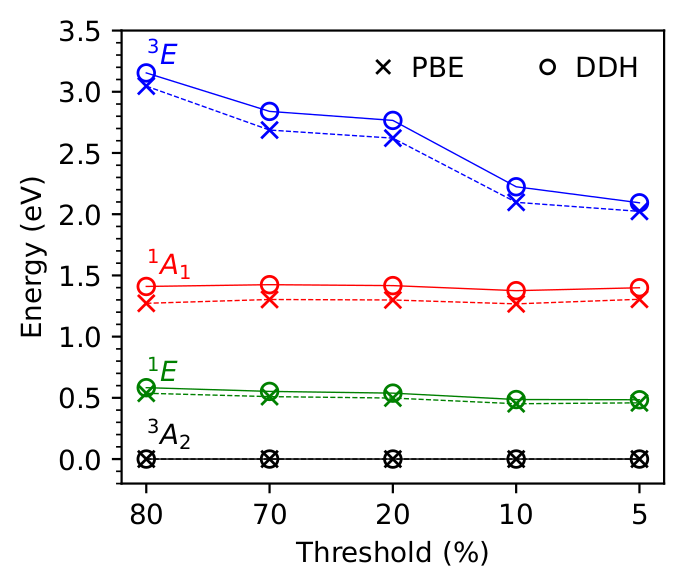
Green's Function Formulation of Quantum Defect Embedding Theory
Nan Sheng, Christian Vorwerk, Marco Govoni, and Giulia Galli
J. Chem. Theory Comput. 18, 6, 3512 (2022). DOI:10.1021/acs.jctc.2c00240
We present a Green's function formulation of the quantum defect embedding theory (QDET) where a double counting scheme is rigorously derived within the G0W0 approximation. We then show the robustness of our methodology by applying the theory with the newly derived scheme to several defects in diamond. Additionally, we discuss a strategy to obtain converged results as a function of the size and composition of the active space. Our results show that QDET is a promising approach to investigate strongly correlated states of defects in solids.
Simulating the Electronic Structure of Spin Defects on Quantum Computers
Benchen Huang, Marco Govoni, and Giulia Galli
We present calculations of the ground and excited state energies of spin defects in solids carried out on a quantum computer, using a hybrid classical/quantum protocol. We focus on the negatively charged nitrogen vacancy center in diamond and on the double vacancy in 4H-SiC, which are of interest for the realization of quantum technologies. We employ a recently developed first-principle quantum embedding theory to describe point defects embedded in a periodic crystal, and to derive an effective Hamiltonian, which is then transformed to a qubit Hamiltonian by means of a parity transformation. We use the variational quantum eigensolver (VQE) and quantum subspace expansion methods to obtain the ground and excited states of spin qubits, respectively, and we propose a promising strategy for noise mitigation. We show that by combining zero-noise extrapolation techniques and constraints on electron occupation to overcome the unphysical state problem of the VQE algorithm, one can obtain reasonably accurate results on near-term-noisy architectures for ground and excited state properties of spin defects.

Photoelectron spectra of water and simple aqueous solutions at extreme conditions
Zifan Ye, Cunzhi Zhang, and Giulia Galli
Faraday Discuss. 236, 352 (2022). DOI:10.1039/D2FD00003B
Determining the electronic structure of aqueous solutions at extreme conditions is an important step towards understanding chemical bonding and reactions in water under pressure (P) and at high temperature (T). We present calculations of the photoelectron spectra of water and a simple solution of NaCl under pressure at conditions relevant to the Earth’s interior (11 GPa and 1000K). We combine first-principles and deep-potential molecular dynamics with electronic structure calculations with dielectric-dependent hybrid functionals. These functionals are defined with a fraction of exact exchange determined from the dielectric constant of the liquid computed in extreme conditions. We find a broadening of the spectra relative to ambient conditions, particularly prominent in the merging of the two main peaks below the onset of the spectra. Furthermore we find an overall red shift at high pressure and temperature, which is however not constant over the whole energy range and varies between 1.1 and 2.4 eV. Our results also show that the anion energy levels are closer to the valence band maximum of the liquid than at ambient conditions, indicating that as P and T are increased, the defect levels of Cl− and OH− in water may eventually lie below the valence band maximum of water. Finally, we characterize the ionization potential of hydrated species deriving from rapid water dissociation, e.g. hydrated hydroxide and hydronium, and we elucidate the electronic states associated with proton transfer events at high pressure. Our results represent a first, important step in predicting the electronic properties of solutions in super-critical conditions.
Active Learning of Polarizable Nanoparticle Phase Diagrams for the Guided Design of Triggerable Self-Assembling Superlattices
Siva Dasetty, Igor Coropceanu, Josh Portner, Jiyuan Li, Juan J. de Pablo, Dmitri Talapin, and Andrew L. Ferguson
Mol. Syst. Des. Eng. 7, 350 (2022).
Polarizable nanoparticles are of interest in materials science because of their rich and complex phase behavior that can be used to engineer nanostructured materials with long-range crystalline order. To understand and rationally navigate the design space of polarizable nanoparticles for self-assembling highly ordered superlattices, we developed a coarse-grained computational model to describe the nanoparticle-nanoparticle interactions in implicit solvent and employ the computationally efficient image method to model many-body polarization interactions. We conducted high-throughput virtual screening over a five-dimensional particle design space spanned by temperature, particle size, particle charge, particle dielectric, and solvent dielectric using enhanced sampling molecular dynamics calculations within an active learning framework to efficiently map out the regions of thermodynamic stability of the self-assembled aggregates. We validate our predictions in comparisons against small angle x-ray scattering measurements of gold nanoparticles surface functionalized with metal chalcogenide ligands. Finally, we use our validated phase maps to computationally design switchable nanostructured materials capable of triggered assembly and disassembly as a function of temperature and solvent dielectric with potential applications as sensors, smart windows, optoelectronic devices, and in medical diagnostics.
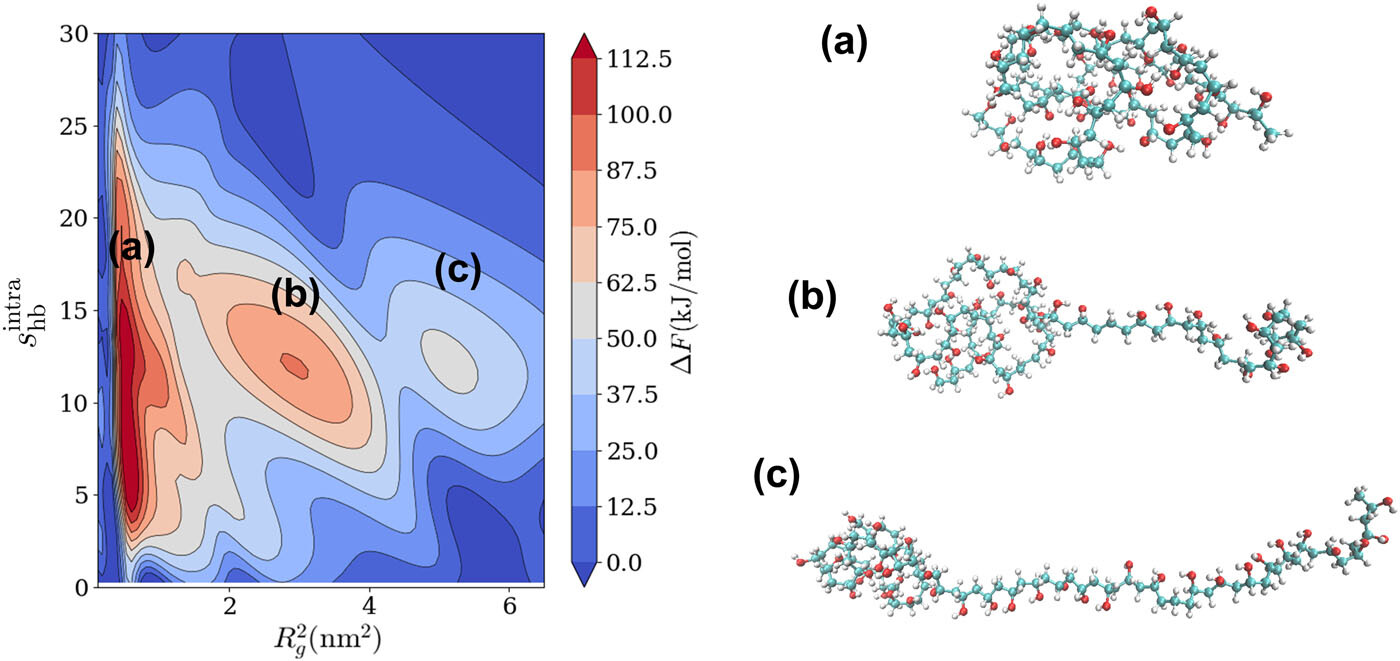
A molecular view of plasticization of polyvinyl alcohol
Ernesto C. Cortés-Morales, Vikramjit S. Rathee, Ahmad Ghobadi, and Jonathan K. Whitmer
J. Chem. Phys. 155, 174903 (2021). DOI:10.1063/5.0065964
Although macromolecules such as polymers are in widespread industrial use, pure formulations rarely have precisely the properties new applications demand. Pure polymer is often too brittle and inflexible, necessitating plasticizers to soften or toughen films and bulk polymer materials. In practice, new formulations are developed by extensive trial-and-error methods, as no general molecular explanations exist for the mechanism of plasticization to aid in determining the optimal structure and concentration of plasticizers. Here, through atomistic molecular simulations augmented with advanced sampling techniques, we develop an atomic-level picture of the processes in plasticization by directly calculating free energies that govern the interaction between polymers and small-molecule plasticizers. This work focuses on the influence of two common plasticizer molecules—glycerol and sorbitol—interacting with polyvinyl alcohol (PVA), a frequently used component of polymer films. In particular, we focus on conformational and hydrogen bond structure changes induced in globules of PVA by the plasticizer molecules, with the hypothesis that hydrogen bonding plays a role in the incorporation of these plasticizers into PVA and, thus, in the observed mechanical properties. While we focus on nanoscopic systems, we observe distinct preferences in the conformational free energy that can be connected to the performance of polymer materials at laboratory and industrial scales. This work presents a new molecular perspective from which effective plasticizers can be developed and presents a firm basis from which important analyses of plasticization in complex chemical environments relevant to industry may be developed.
Stability and Molecular Pathways to the Formation of Spin Defects in Silicon Carbide
Elizabeth M. Y. Lee, Alvin Yu, Juan J. de Pablo, and Giulia Galli
Nature Communications 12, 6325 (2021). DOI:10.1038/s41467-021-26419
Spin defects in wide-bandgap semiconductors provide a promising platform to create qubits for quantum technologies. Their synthesis, however, presents considerable challenges, and the mechanisms responsible for their generation or annihilation are poorly understood. Here, we elucidate spin defect formation processes in a binary crystal for a key qubit candidate—the divacancy complex (VV) in silicon carbide (SiC). Using atomistic models, enhanced sampling simulations, and density functional theory calculations, we find that VV formation is a thermally activated process that competes with the conversion of silicon (VSi) to carbon monovacancies (VC), and that VV reorientation can occur without dissociation. We also find that increasing the concentration of VSi relative to VC favors the formation of divacancies. Moreover, we identify pathways to create spin defects consisting of antisite-double vacancy complexes and determine their electronic properties. The detailed view of the mechanisms that underpin the formation and dynamics of spin defects presented here may facilitate the realization of qubits in an industrially relevant material.
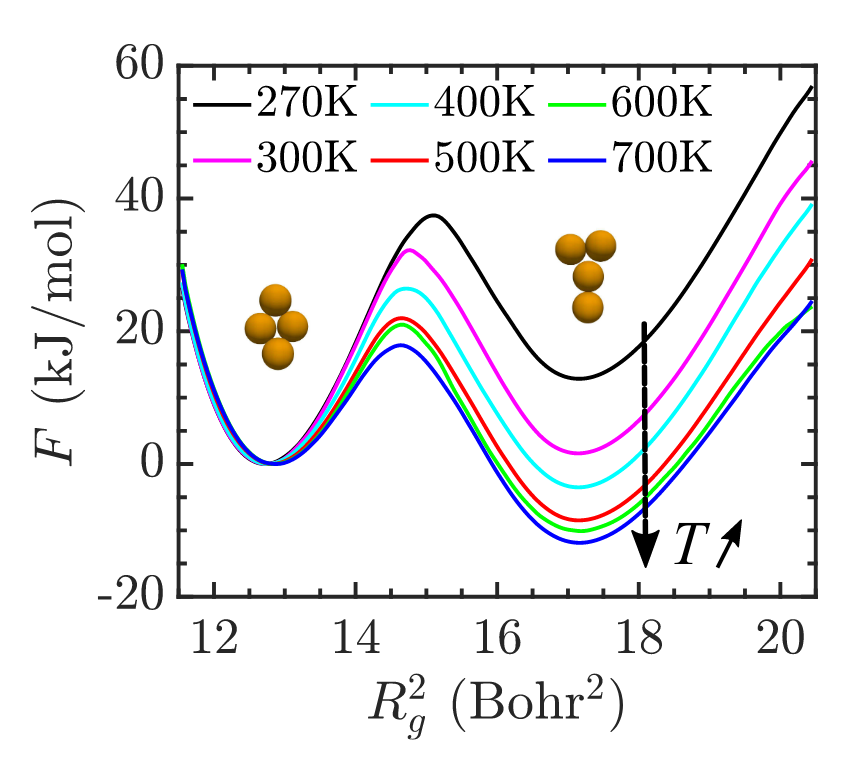
Free Energy Landscape and Isomerization Rates of Au4 Clusters at Finite Temperature
Jiale Shi, Shanghui Huang, Francois Gygi, and Jonathan K. Whitmer
J. Phys. Chem. A 126, 21, 3392 (2022). DOI:10.1021/acs.jpca.2c02732
In metallic nanoparticles, the cluster geometric structures control the particle’s electronic band structure, polarizability, and catalytic properties. Analyzing the structural properties is a complex problem; the structure of an assembled cluster changes from moment to moment due to thermal fluctuations. Conventional structural analyses based on spectroscopy or diffraction cannot determine the instantaneous structure exactly and canmerely provide an averaged structure. Molecular simulations offer an opportunity to examine the assembly and evolution of metallic clusters, as the preferred assemblies and conformations can easily be visualized and explored. Here, we utilize the adaptive biasing force algorithm applied to first principles molecular dynamicsto demonstrate exploration of a relatively simple system which permits comprehensive study of the small metal cluster Au4 in both neutral and charged configurations. Our simulation work offers a quantitative understanding of these clusters’ dynamic structure, which is significant for single-site catalytic reactions on metal clusters and provides a starting point for a detailed quantitative understanding of more complex pure metal and alloy clusters’ dynamic properties.
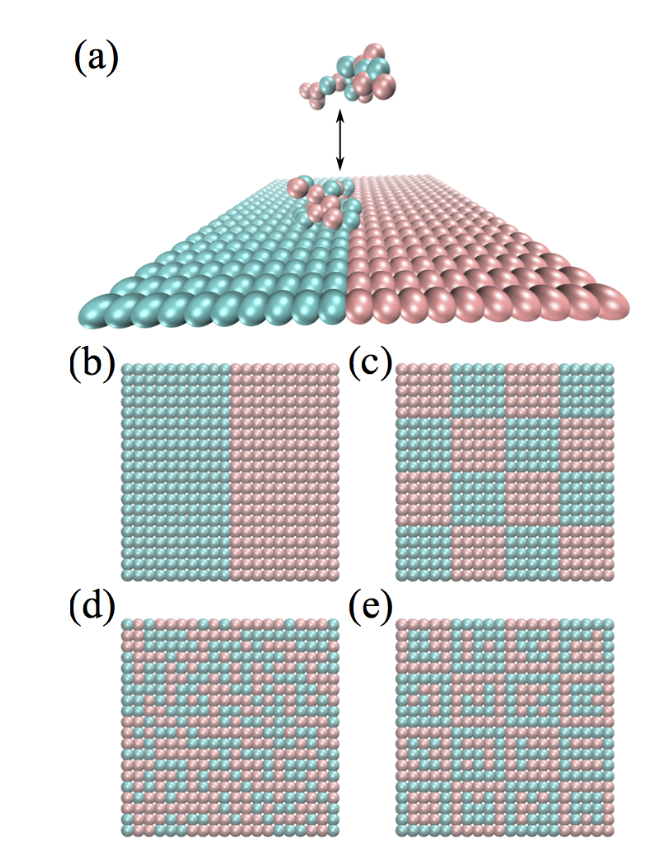
Predicting Adhesive Free Energies of Polymer-Surface Interactions with Machine Learning
Jiale Shi, Michael J. Quevillon, Pedro H. Amorim Valenca, and Jonathan K. Whitmer
ACS Appl. Mater. Interfaces 14, 32, 37161 (2022). DOI:10.1021/acsami.2c08891
Polymer-surface interactions are crucial to many biological processes and industrial applications. Here we propose a machine-learning method to connect a modelpolymer’s sequence with its adhesion to decorated surfaces. We simulate the adhesive free energies of 20,000 unique coarse-grained 1D sequential polymers interacting with functionalized surfaces and build support vector regression (SVR) models that demonstrate inexpensive and reliable prediction of the adhesive free energy as a functionof the sequence. Our work highlights the promising integration of coarse-grained simulation with data-driven machine learning methods for the design of new functional polymers and represents an important step toward linking polymer compositions with polymer-surface interactions.
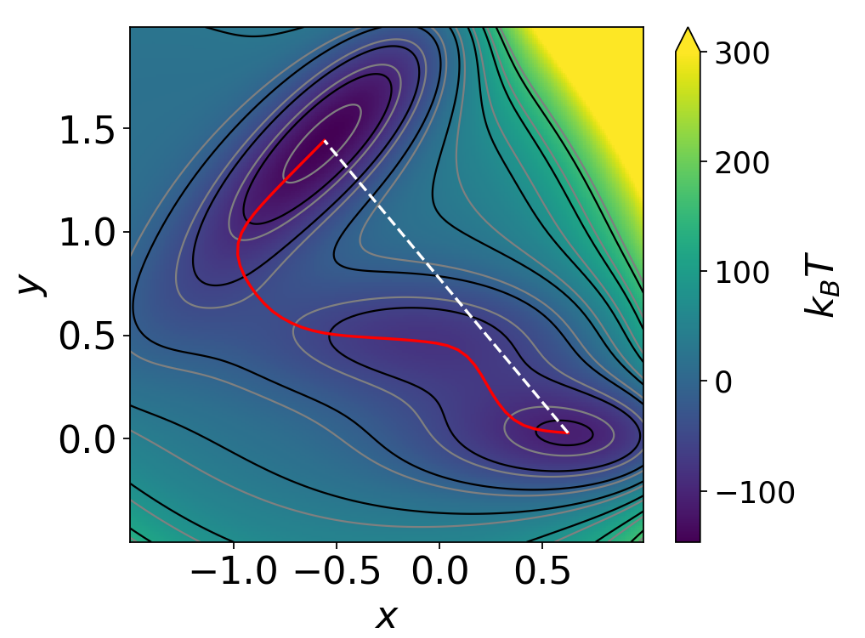
Exploring the Potential of Parallel Biasing in Flat Histogram Methods
Shanghui Huang, Michal J. Quevillon, Ernesto C. Cortes-Morales, and Jonathan K. Whitmer
Submitted (2021). arXiv:2109.05005
Metadynamics, a member of the 'flat histogram' class of advanced sampling algorithms, has been widely used in molecular simulations to drive the exploration of states separated by high free energy barriers and promote comprehensive sampling of free energy landscapes defined on collective variables (CVs) which characterize the state of the system. Typically, the methods encounter severe limitations when exploring large numbersof CVs. A recently proposed variant, parallel bias metadynamics(PBMetaD), promises to aid in exploring free energy landscapes along with multiple important collective variables by exchanging then-dimensional free energy landscape required by standardmethods for none-dimensional marginal free energy landscapes. In thisstudy, we systematically examine how parallel biasing affects the convergence of free energy landscapes along with each variable relative to standard methods and the effectiveness of the parallel biasing strategy for addressing common bottlenecks in the use of advanced sampling to calculate free energies.
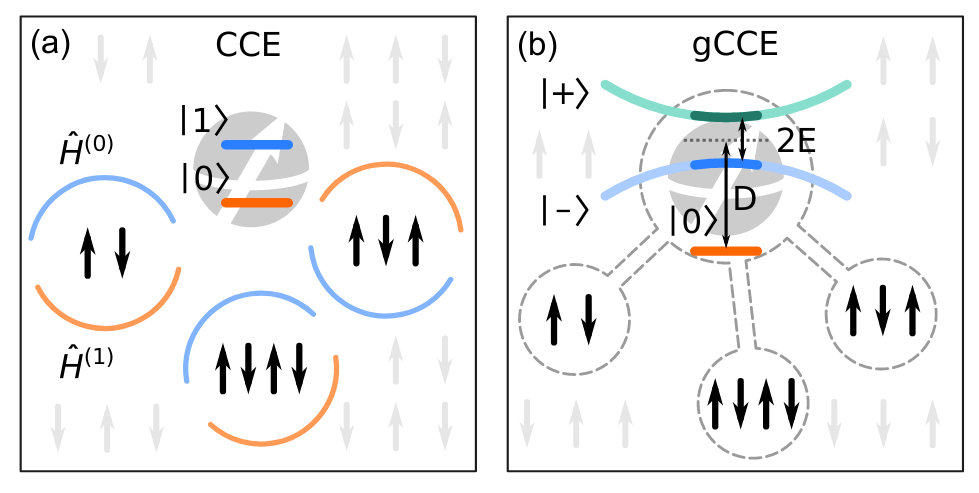
PyCCE: A Python Package for Cluster Correlation Expansion Simulations of Spin Qubit Dynamics
Mykyta Onizhuk and Giulia Galli
Adv. Theory Simul. 4: 2100254 (2021). DOI: 10.1002/adts.202100254
We present PyCCE, an open-source Python library to simulate the dynamics of spin qubits in a spin bath, using the cluster-correlation expansion (CCE) method. PyCCE includes modules togenerate realistic spin baths, employing coupling parameters computed from first principles with electronic structure codes, and enables the user to run simulations with either the conventional or generalized CCE method. We illustrate three use cases of the Python library: the calculation of the Hahn-echo coherence time of the nitrogen vacancy in diamond; the calculation of the coherence tim eof the basal di-vacancy in silicon carbide at avoided crossings; and the magnetic field orientation-dependent dynamics of a shallow donor in silicon. The complete documentation and installation instructions are available at https://pycce.readthedocs.io/en/latest/
Combined first-principles calculations of electron-electron and electron-phonon self-energies in condensed systems
Han Yang, Marco Govoni, Arpan Kundu, and Giulia Galli
J. Chem. Theory Comput. 17, 12, 7468 (2021). DOI:10.1021/acs.jctc.1c00605
We present a method to efficiently combine the computation of electron-electron andelectron-phonon self-energies, which enables the evaluation of electron-phonon coupling at the G0W0 level of theory for systems with hundreds of atoms. In addition, our approach, which is a generalization of a method recently proposed for molecules [J.Chem. Theory Comput. 2018, 14, 6269–6275], enables the inclusion of non-adiabaticand temperature effects at no additional computational cost. We present results for diamond and defects in diamond and discuss the importance of numerically accurate G0W0 band structures to obtain robust predictions of zero point renormalization (ZPR)of band gaps, and of the inclusion of non-adiabatic effect to accurately compute the ZPR of defect states in the band gap.
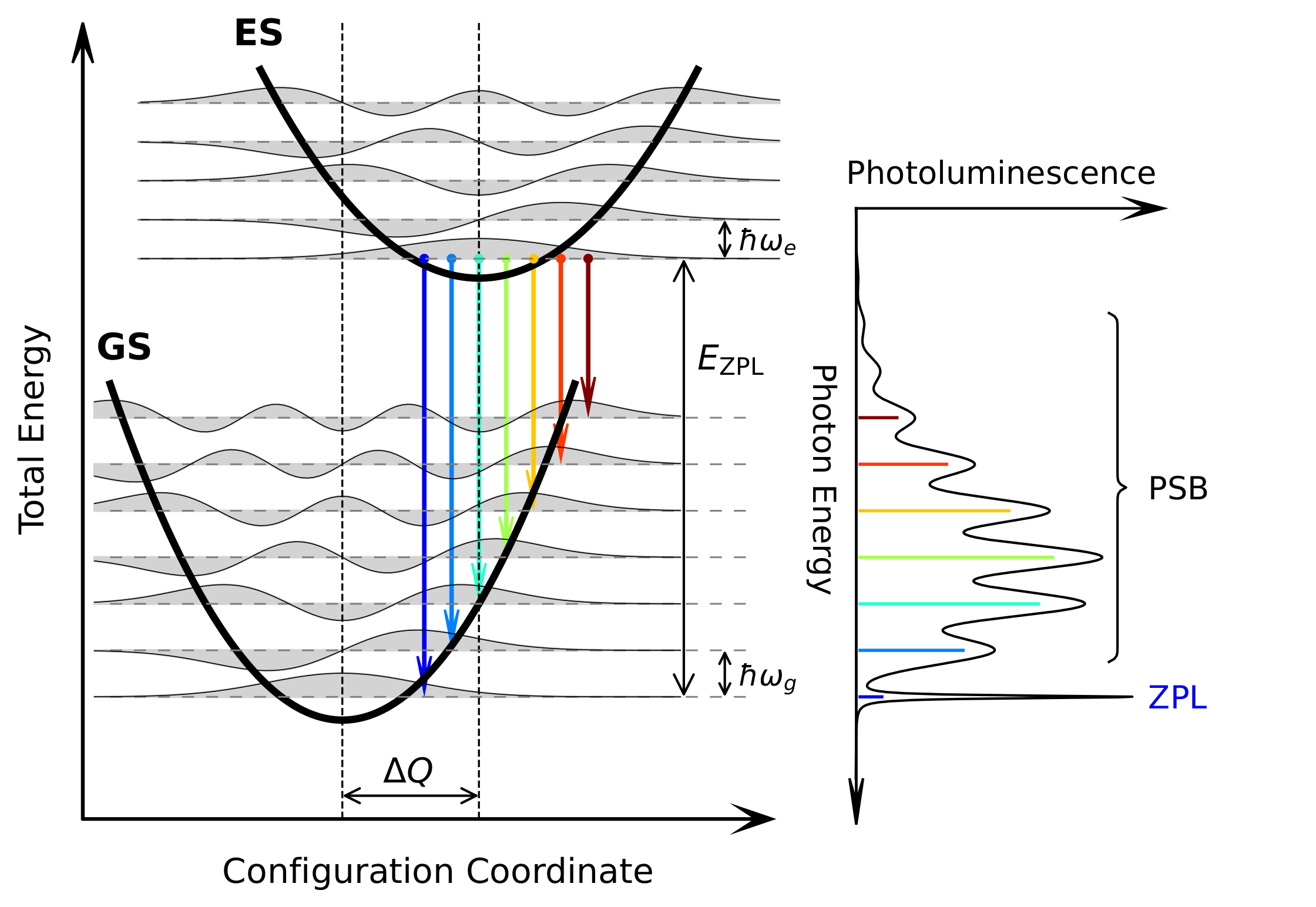
Photoluminescence spectra of point defects in semiconductors: validation of first principles calculations
Yu Jin, Marco Govoni, Gary Wolfowicz, Sean E. Sullivan, F. Joseph Heremans, David D. Awschalom, and Giulia Galli
Phys. Rev. Materials 5, 084603 (2021). DOI: 10.1103/PhysRevMaterials.5.084603
Optically and magnetically active point defects in semiconductors are interesting platforms for the development of solid-state quantum technologies. Their optical properties are usually probed by measuring photoluminescence spectra, which provide information on excitation energies and on the interaction of electrons with lattice vibrations. We present a combined computational and experimental study of photoluminescence spectra of defects in diamond and SiC, aimed at assessing the validity of theoretical and numerical approximations used in first principles calculations, including the use of the Franck-Condon principle and the displaced harmonic oscillator approximation. We focus on prototypical examples of solid-state qubits, the divacancy centers in SiC and the nitrogen-vacancy in diamond, and we report computed photoluminescence spectra as a function of temperature that are in very good agreement with the measured ones. As expected we find that the use of hybrid functionals leads to more accurate results than semilocal functionals. Interestingly our calculations show that constrained density functional theory (CDFT) and time-dependent hybrid DFT perform equally well in describing the excited state potential energy surface of triplet states; our findings indicate that CDFT, a relatively cheap computational approach, is sufficiently accurate for the calculations of photoluminescence spectra of the defects studied here. Finally, we find that only by correcting for finite-size effects and extrapolating to the dilute limit, one can obtain a good agreement between theory and experiment. Our results provide a detailed validation protocol of first principles calculations of photoluminescence spectra, necessary both for the interpretation of experiments and for robust predictions of the electronic properties of point defects insemiconductors.
Quantum Embedding Theories to Simulate Condensed Systems on Quantum Computers
Christian Vorwerk, Nan Sheng, Marco Govoni, Benchen Huang, and Giulia Galli
Nature Computational Science 2, 424 (2022). DOI: 10.1038/s43588-022-00279-0
We discuss computational frameworks to carry out electronic structure calculations of materials on noisy intermediate scale quantum computers using embedding theories and effective manybody Hamiltonians. The latter are defined and diagonalized by performing hybrid computations on classical and quantum architectures. We present applications for a specific class of materials, i.e., spin-defects in solids, which are promising systems to build future quantum technologies, e.g., computers, sensors and devices for quantum communications.
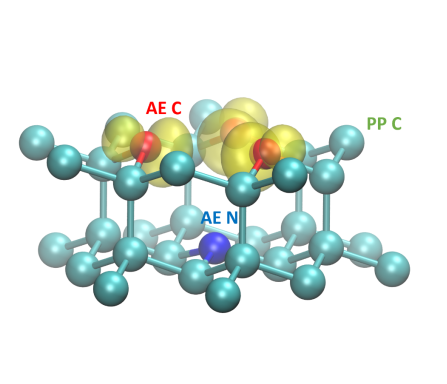
Spin-spin interactions in solids from mixed all-electron and pseudopotential calculations—a path to screening materials for spin qubits
Krishnendu Ghosh, He Ma, Mykyta Onizhuk, Vikram Gavini, Giulia Galli
npj Computational Materials 7, 123 (2021). DOI: 10.1038/s41524-021-00590-w
Understanding the quantum dynamics of spin defects and their coherence properties requires an accurate modeling of spin-spin interaction in solids and molecules, for example by using spin Hamiltonians with parameters obtained from first-principles calculations. We present a real-space approach based on density functional theory for the calculation of spin-Hamiltonian parameters, where only selected atoms are treated at the all-electron level, while the rest of the system is described with the pseudopotential approximation. Our approach permits calculations for systems containing more than 1000 atoms, as demonstrated for defects in diamond and silicon carbide. We show that only a small number of atoms surrounding the defect needs to be treated at the all-electron level, in order to obtain an overall all-electron accuracy for hyperfine and zero-field splitting tensors. We also present results for coherence times, computed with the cluster correlation expansion method, highlighting the importance of accurate spin-Hamiltonian parameters for quantitative predictions of spin dynamics.
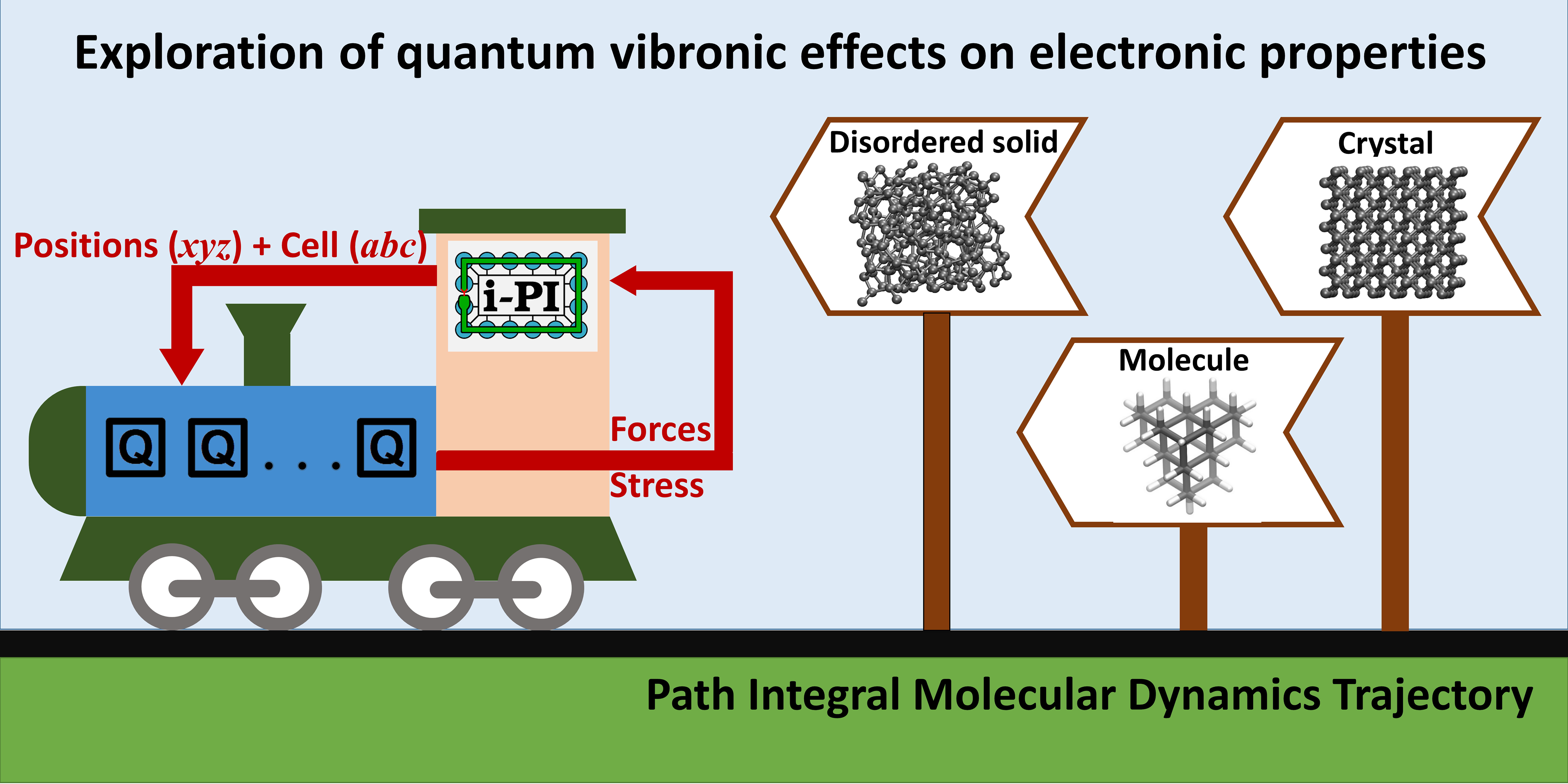
Quantum Vibronic Effects on the Electronic Properties of Solid and Molecular Carbon
Arpan Kundu, Marco Govoni, Han Yang, Michele Ceriotti, Francois Gygi, and Giulia Galli
Phys. Rev. Materials 5, L070801 (2021). DOI: 10.1103/PhysRevMaterials.5.L070801
We study the effect of quantum vibronic coupling on the electronic properties of carbon allotropes, including molecules and solids, by combining path integral first principles molecular dynamics (FPMD) with a colored noise thermostat. In addition to avoiding several approximations commonly adopted in calculations of electron-phonon coupling, our approach only adds a moderate computational cost to FPMD simulations and hence it is applicable to large supercells, such as those required to describe amorphous solids. We predict the effect of electron-phonon coupling on the fundamental gap of amorphous carbon, and we show that in diamond the zero-phonon renormalization of the band gap is larger than previously reported.
OPTIMADE: an API for exchanging materials data
Casper W. Andersen, Rickard Armiento, Evgeny Blokhin, Gareth J. Conduit, Shyam Dwaraknath, Matthew L. Evans, Adam Fekete, Abhijith Gopakumar, Saulius Grazulis, Andrius Merkys, Fawzi Mohamed, Corey Oses, Giovanni Pizzi, Gian-Marco Rignanese, Markus Scheidgen, Leopold Talirz, Cormac Toher, Donald Winston, Rossella Aversa, Kamal Choudhary, Pauline Colinet, Stefano Curtarolo, Davide Di Stefano, Claudia Draxl, Suleyman Er, Marco Esters, Marco Fornari, Matteo Giantomassi, Marco Govoni, Geoffroy Hautier, Vinay Hegde, Matthew K. Horton, Patrick Huck, Georg Huhs, Jens Hummelshøj, Ankit Kariryaa, Boris Kozinsky, Snehal Kumbhar, Mohan Liu, Nicola Marzari, Andrew J. Morris, Arash Mostofi, Kristin A. Persson, Guido Petretto, Thomas Purcell, Francesco Ricci, Frisco Rose, Matthias Scheffler, Daniel Speckhard, Martin Uhrin, Antanas Vaitkus, Pierre Villars, David Waroquiers, Chris Wolverton, Michael Wu, and Xiaoyu Yang
Scientific Data 8, 217 (2021). DOI: 10.1038/s41597-021-00974-z
The Open Databases Integration for Materials Design (OPTIMADE) consortium has designed a universal application programming interface (API) to make materials databases accessible and interoperable. We outline the first stable release of the specification, v1.0, which is already supported by many leading databases and several software packages. We illustrate the advantages of the OPTIMADE API through worked examples on each of the public materials databases that support the full API specification.

Semiconductor quantum dots: Technological progress and future challenges
F. Pelayo García de Arquer, Dmitri Talapin, Victor I. Klimov, Yasuhiko Arakawa, Manfred Bayer, and Edward Sargent
Science 373, eaaz8541 (2021). DOI: 10.1126/science.aaz8541
Semiconductor materials feature optical and electronic properties that can be engineered through their composition and crystal structure. The use of semiconductors such as silicon gallium arsenide sparked technologies from computers and mobile phones to lasers and satellites. Semiconductor quantum dots (QDs) offer an additional lever: Because their size is reduced to the nanometer scale in all three dimensions, the restricted electron motion leads to a discrete atom-like electronic structure and size-dependent energy levels. This enables the design of nanomaterials with widely tunable light absorption, bright emission of pure colors, control over electronic transport, and a wide tuning of chemical and physical functions because of their large surface-to-volume ratio.
Transformation between elastic dipoles, quadrupoles, octupoles, and hexadecapoles driven by surfactant self-assembly in nematic emulsion
Bohdan Senyuk, Ali Mozaffari, Kevin Crust, Rui Zhang, Juan J. de Pablo, and Ivan I. Smalyukh
Sci. Adv. 7, eabg0377 (2021). DOI: 10.1126/sciadv.abg0377
Emulsions comprising isotropic fluid drops within a nematic host are of interest for applications ranging from biodetection to smart windows, which rely on changes of molecular alignment structures around the drops in response to chemical, thermal, electric, and other stimuli. We show that absorption or desorption of trace amounts of common surfactants can drive continuous transformations of elastic multipoles induced by the droplets within the uniformly aligned nematic host. Out-of-equilibrium dynamics of director structures emerge from a controlled self-assembly or desorption of different surfactants at the drop-nematic interfaces, with ensuing forward and reverse transformations between elastic dipoles, quadrupoles, octupoles, and hexadecapoles. We characterize intertransformations of droplet-induced surface and bulk defects, probe elastic pair interactions, and discuss emergent prospects for fundamental science and applications of the reconfigurable nematic emulsions.
Anisotropic Coarse-Grained Model for Conjugated Polymers: Investigations into Solution Morphologies
Alexander E. Cohen, Nicholas E. Jackson, and Juan J. de Pablo
Macromolecules, 54, 3780-3789 (2021). DOI: 10.1021/acs.macromol.1c00302
The optoelectronic properties of conjugated polymers are dictated by the interplay of multiscale structural features, including intrachain dihedrals, interchain π–π stacking, and the complex mesoscale morphology. While much is known about the structures of polymers with isotropically interacting monomers, little is known about polymers with strongly anisotropic backbone monomers, a class of materials to which conjugated polymers belong. Fundamental understanding is further complicated when the semiflexible and molecularly heterogeneous nature of conjugated polymers is taken into account. We present an anisotropic coarse-grained (CG) model for conjugated polymers that incorporates key molecular features (monomer anisotropy, intermonomer dihedrals, and side chains). The model is employed to characterize the single-chain conformational properties of conjugated polymers, revealing a rich temperature-dependent conformational landscape. These studies provide a critical link between CG molecular descriptors and conformational ordering in conjugated polymers.
Quantum Embedding Theory for Strongly-correlated States in Material
He Ma, Nan Sheng, Marco Govoni, and Giulia Galli
J. Chem. Theory Comput., 17, 2116-2125 (2021). DOI: 10.1021/acs.jctc.0c01258
Quantum embedding theories are promising approaches to investigate stronglycorrelated electronic states of active regions of large-scale molecular or condensed systems. Notable examples are spin defects in semiconductors and insulators. We present a detailed derivation of a quantum embedding theory recently introduced, which is based on the definition of effective Hamiltonians. The effect of the environment on a chosen active space is accounted for through screened Coulomb interactions evaluated using density functional theory. Importantly, the random phase approximation is not required and the evaluation of virtual electronic orbitals is circumvented with algorithms previously developed in the context of calculations based on many-body perturbation theory. In addition, we generalize the quantum embedding theory to active spaces composed of orbitals that are not eigenstates of Kohn-Sham Hamiltonians. Finally, we report results for spin defects in semiconductors.
Neural Network Sampling of the Free Energy Landscape for Nitrogen Dissociation on Ruthenium
Elizabeth M. Y. Lee, Thomas Ludwig, Boyuan Yu, Aayush R. Singh, François Gygi, Jens K. Nørskov, and Juan J. de Pablo
J. Phys. Chem. Lett. 12, 2954-2962, (2021). DOI: 10.1021/acs.jpclett.1c00195
In heterogeneous catalysis, free energy profiles of reactions govern the mechanisms, rates, and equilibria. Energetics are conventionally computed using the harmonic approximation (HA), which requires the determination of critical states a priori. Here, we use neural networks to efficiently sample and directly calculate the free energy surface (FES) of a prototypical heterogeneous catalysis reaction—the dissociation of molecular nitrogen on ruthenium—at density functional theory-level accuracy. We find that the vibrational entropy of surface atoms, often neglected in HA for transition metal catalysts, contributes significantly to the reaction barrier. The minimum free energy path for dissociation reveals an “on-top” adsorbed molecular state prior to the transition state. While a previously reported flat-lying molecular metastable state can be identified in the potential energy surface, it is absent in the FES at relevant reaction temperatures. These findings demonstrate the importance of identifying critical points self-consistently on the FES for reactions that involve considerable entropic effects.
Machine Learning Dielectric Screening for the Simulation of Excited State Properties of Molecules and Materials
Sijia Dong, Marco Govoni and Giulia Galli
Chem. Sci. 12, 4970-4980 (2021). DOI: 10.1039/D1SC00503K
Accurate and efficient calculations of absorption spectra of molecules and materials at finite temperature are essential for the understanding and rational design of broad classes of systems. Solving the Bethe-Salpeter equation (BSE) for electron-hole pairs usually yields accurate predictions of absorption spectra, but it is computationally expensive, especially if calculations for multiple configurations at finite temperature are required. We present an approach to improve the efficiency of first principles calculations of absorption spectra at finite temperature, based on the solution of the BSE in finite electric field and machine-learning techniques. We demonstrate that methods based on convolutional neural networks can be efficiently used to compute the screened Coulomb interaction, a key component of BSE calculations, with computational gains of one to two orders of magnitude for systems with 50 to 500 atoms, including liquids, solids, nanostructures, and solid/liquid interfaces. Importantly, our approach yields interpretable machine learning results, from which model dielectric functions may be derived. These model functions may be used not only in the BSE but also in developing functionals for time-dependent density functional theory (TDDFT) calculations, for both homogeneous and heterogeneous systems. Our work provides a strategy to combine machine learning with electronic structure calculations to accelerate the first principles simulations of excited-state properties.
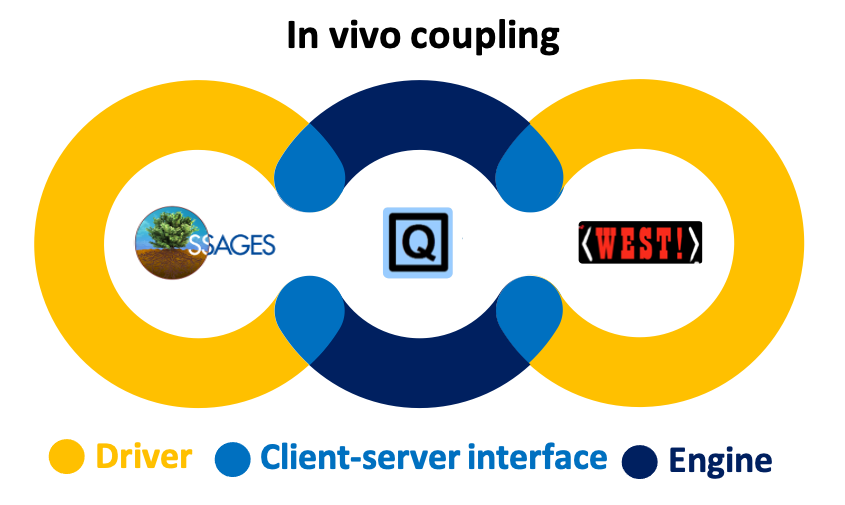
Code interoperability extends the scope of quantum simulations
Marco Govoni, Jonathan Whitmer, Juan de Pablo, Francois Gygi, and Giulia Galli
npj Comput. Mater. 7, 32, (2021). DOI: 10.1038/s41524-021-00501-z.
The functionality of many materials is critically dependent on the integration of dissimilar components and on the interfaces that arise between them. The description of such heterogeneous components requires the development and deployment of first principles methods, coupled to appropriate dynamical descriptions of matter and advanced sampling techniques, in order to capture all the relevant length and time scales of importance to the materials’ performance. It is thus essential to build simple, streamlined computational schemes for the prediction and design of multiple properties of broad classes of materials, by developing interoperable codes which can be efficiently coupled to each other to perform complex tasks. We discuss the use of interoperable codes to simulate the structural and spectroscopic characterization of materials, including chemical reactions for catalysis, the description of defects for quantum information science, and heat and charge transport.
Determining the Structure–Property Relationships of Quasi-Two-Dimensional Semiconductor Nanoplatelets
Arin R. Greenwood, Sergio Mazzotti, David J. Norris, and Giulia Galli
J. Phys. Chem. C 125, 8, 4820-4827 (2021). DOI: 10.1021/acs.jpcc.0c10559.
We report a theoretical study of CdSe nanoplatelets aimed at identifying the main factors determining their photophysical properties. Using atomic configurations optimized with density functional theory calculations, we computed quasiparticle and exciton binding energies of nanoplatelets with two to seven monolayers. We employed many body perturbation theory at the GW level and solved the Bethe-Salpeter equation to obtain absorption spectra and excitonic properties. Our results, which agree well with recent experiments, were then used to design a model that allows us to disentangle the effects of quantum confinement, strain induced by passivating ligands, and dielectric environment on the electronic properties of nanoplatelets. We found that, for the model to accurately reproduce our first principle results, it is critical to account for surface stress and consider a finite potential barrier and energy-dependent effective masses when describing quantum confinement. Our findings call into question previous assumptions on the validity of an infinite barrier to describe carrier confinement in nanoplatelets, suggesting that it may be possible to optimize interfacial charge transfer and extraction by appropriately choosing passivating ligands. The model developed here is generalizable to core–shell platelets and enables the description of system sizes not yet directly treatable by first-principles calculations.
Targeted sequence design within the coarse-grained polymer genome
Michael A. Webb, Nicholas E. Jackson, Phwey S. Gil, and Juan J. de Pablo
Sci. Adv. 6, 43, eabc6216 (2020). DOI: 10.1126/sciadv.abc6216
The chemical design of polymers with target structural and/or functional properties represents a grand challenge in materials science. While data-driven design approaches are promising, success with polymers has been limited, largely due to limitations in data availability. Here, we demonstrate the targeted sequence design of single-chain structure in polymers by combining coarse-grained modeling, machine learning, and model optimization. Nearly 2000 unique coarse-grained polymers are simulated to construct and analyze machine learning models. We find that deep neural networks inexpensively and reliably predict structural properties with limited sequence infor-mation as input. By coupling trained ML models with sequential model-based optimization, polymer sequences are proposed to exhibit globular, swollen, or rod-like behaviors, which are verified by explicit simulations. This work highlights the promising integration of coarse-grained modeling with data-driven design and represents a necessary and crucial step toward more complex polymer design efforts.

Validating first-principles molecular dynamics calculations of oxide/water interfaces with x-ray reflectivity data
Katherine J. Harmon, Kendra Letchworth-Weaver, Alex P. Gaiduk, Federico Giberti, Maria Chan, Francois Gygi, Paul Fenter and Giulia Galli
Phys. Rev. Mat. 4, 113805 (2020). DOI: 10.1103/PhysRevMaterials.4.113805
Metal oxide/water interfaces play a crucial role in many electrochemical and photocatalytic processes, such as photoelectrochemical water splitting, the creation of fuel from sunlight, and electrochemical CO2 reduction. First-principles electronic structure calculations can reveal unique insights into these processes, such as the role of the alignment of the oxide electronic energy levels with those of liquid water. An essential prerequisite for the success of such calculations is the ability to predict accurate structural models of these interfaces, which in turn requires careful experimental validation. Here we report a general, quantitative validation protocol for first-principles molecular dynamics simulations of oxide/aqueous interfaces. The approach makes direct comparisons of interfacial x-ray reflectivity (XR) signals from experimental measurements and those obtained from ab initio simulations with semilocal and van der Waals functionals. The protocol is demonstrated here for the case of the Al2O3(001)/water interface, one of the simplest oxide/water interfaces. We discuss the technical requirements needed for validation, including the choice of the density functional, the simulation cell size, and the optimal choice of the thermodynamic ensemble. Our results establish a general paradigm for the validation of structural models and interactions at solid/water interfaces derived from first-principles simulations. While there is qualitative agreement between the simulated structures and the experimental best-fit structure, direct comparisons of simulated and measured XR intensities show quantitative discrepancies that derive from both bulk regions (i.e., alumina and water) as well as the interfacial region, highlighting the need for accurate density functionals to properly describe interfacial interactions. Our results show that XR data are sensitive not only to the atomic structure (i.e., the atom locations) but also to the electron-density distributions in both the substrate and at the interface.
Stoichiometry of the Core Determines the Electronic Structure of Core-Shell III–V/II–VI Nanoparticles
Mariami Rusishvili, Stefan Wippermann, Dmitri Talapin, and Giulia Galli
Chem. Mater. 32, 22, 9798-9804 (2020). DOI: 10.1021/acs.chemmater.0c03939
Recently III-V quantum dots (QDs) emerged as an environmentally friendly alternative to CdSe, however they exhibit broader emission spectra and inferior photoluminescence quantum yield. Here we report a computational study of the opto-electronic properties of In x P z and In x Ga y P z QDs interfaced with zinc chalcogenide shells. Using density functional theory, we show that fine-tuning the dot composition is critical to achieving narrow emission lines. We show that cores with the same diameter but different stoichiometries may absorb and emit at different wavelengths, thus leading to broad bands, with widths decreasing as the diameter of the dot increases. In particular the value of the fundamental gap depends on the ratio between the number of group III and P atoms and is maximized for 1:1 composition. We also show that the interplay between quantum confinement and strain determines the difference in the electronic properties of III-V QDs interfaced with ZnS or ZnSe shells.
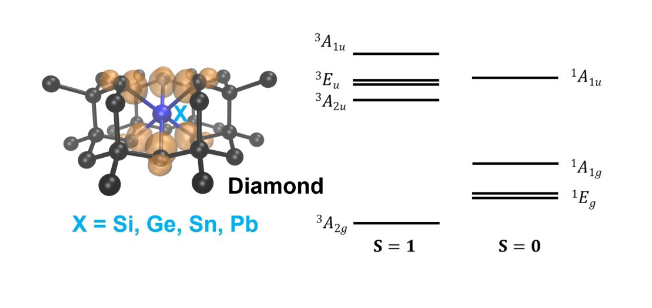
First-principles Studies of Strongly Correlated States in Defect Spin Qubits in Diamond
He Ma, Nan Sheng, Marco Govoni, and Giulia Galli
Phys. Chem. Chem. Phys. 22, 25522-25527 (2020). DOI: 10.1039/D0CP04585C
Using a recently developed quantum embedding theory, we present first principles calculations of strongly correlated states of spin defects in diamond. Within this theory, effective Hamiltonians are constructed, which can be solved by classical and quantum computers; the latter promise a much more favorable scaling as a function of system size than the former. In particular, we report a study of the neutral group-IV vacancy complexes in diamond, and we discuss their strongly-correlated spin-singlet and spin-triplet excited states. Our results provide valuable predictions for experiments aimed at optical manipulation of these defects for quantum information technology applications.
Molecular Latent Space Simulators
Hythem Sidky, Wei Chen and Andrew L. Ferguson
Chem. Sci. 11, 9459-9467 (2020). DOI: 10.1039/D0SC03635H
Small integration time steps limit molecular dynamics (MD) simulations to millisecond time scales. Markov state models (MSMs) and equation-free approaches learn low-dimensional kinetic models from MD simulation data by performing configurational or dynamical coarse-graining of the state space. The learned kinetic models enable the efficient generation of dynamical trajectories over vastly longer time scales than are accessible by MD, but the discretization of configurational space and/or absence of a means to reconstruct molecular configurations precludes the generation of continuous all-atom molecular trajectories. We propose latent space simulators (LSS) to learn kinetic models for continuous all-atom simulation trajectories by training three deep learning networks to (i) learn the slow collective variables of the molecular system, (ii) propagate the system dynamics within this slow latent space, and (iii) generatively reconstruct molecular configurations. We demonstrate the approach in an application to Trp-cage miniprotein to produce novel ultra-long synthetic folding trajectories that accurately reproduce all-atom molecular structure, thermodynamics, and kinetics at six orders of magnitude lower cost than MD. The dramatically lower cost of trajectory generation enables greatly improved sampling and greatly reduced statistical uncertainties in estimated thermodynamic averages and kinetic rates.

Dissociation of salts in water under pressure
Cunzhi Zhang, Federico Giberti, Emre Sevgen, Juan de Pablo, Francois Gygi and Giulia Galli
Nat. Commun. 11, 3037 (2020). DOI: 10.1038/s41467-020-16704-9
The investigation of salts in water at extreme conditions is crucial to understanding the properties of aqueous fluids in the Earth. We report first principles (FP) and classical molecular dynamics simulations of NaCl in the dilute limit, at temperatures and pressures relevant to the Earth’s upper mantle. Similar to ambient conditions, we observe two metastable states of the salt: the contact (CIP) and the solvent-shared ion-pair (SIP), which are entropically and enthalpically favored, respectively. We find that the free energy barrier between the CIP and SIP minima increases at extreme conditions, and that the stability of the CIP is enhanced in FP simulations, consistent with the decrease of the dielectric constant of water. The minimum free energy path between the CIP and SIP becomes smoother at high pressure, and the relative stability of the two configurations is affected by water self-dissociation, which can only be described properly by FP simulations.
Automated determination of n-cyanobiphenyl and n-cyanobiphenyl binary mixtures elastic constants in the nematic phase from molecular simulation
Jiale Shi, Hythem Sidky, and Jonathan K. Whitmer
Mol. Syst. Des. Eng. 5, 1131-1136, (2020)
New applications of liquid crystalline materials have increased the need for precise engineering of elastic properties. Recently, Sidky et al. [H. Sidky, J. J. de Pablo, and J. K. Whitmer Phys. Rev. Lett., 2018, 120, 107801] presented methods by which the elastic coefficients of molecular models with atomistic detail can be accurately calculated, demonstrating the result for the ubiquitous mesogen 5CB. However, it is beneficial to future applications to demonstrate how the process may be refined into a precise tool for engineering functional materials. In this work, these techniques are applied to the homologous series of nCB materials and their binary mixtures, focusing on the standard bend, twist, and splay deformations, using an automated process. Our results show exceptional agreement with published experimental measurements for the nCBs and present a path forward to computational molecular engineering of liquid crystal elasticity for novel molecules and mixtures.
Quantum simulations of materials on near-term quantum computers
He Ma, Marco Govoni, and Giulia Galli
npj Comput. Mat. 6, 85 (2020). DOI: 10.1038/s41524-020-00353-z
Quantum computers hold promise to enable efficient simulations of the properties of molecules and materials; however, at present they only permit ab initio calculations of a few atoms, due to a limited number of qubits. In order to harness the power of near-term quantum computers for simulations of larger systems, it is desirable to develop hybrid quantum-classical methods where the quantum computation is restricted to a small portion of the system. This is of particular relevance for molecules and solids where an active region requires a higher level of theoretical accuracy than its environment. Here we present a quantum embedding theory for the calculation of strongly-correlated electronic states of active regions, with the rest of the system described within density functional theory. We demonstrate the accuracy and effectiveness of the approach by investigating several defect quantum bits in semiconductors that are of great interest for quantum information technologies. We perform calculations on quantum computers and show that they yield results in agreement with those obtained with exact diagonalization on classical architectures, paving the way to simulations of realistic materials on near-term quantum computers.
PyCDFT: A Python package for constrained density functional theory
He Ma, Wennie Wang, Siyoung Kim, Man-Hin Cheng, Marco Govoni, and Giulia Galli
J. Comp. Chem. 41, 1859-1867 (2020). DOI: 10.1002/jcc.26354
We present PyCDFT, a Python package to compute diabatic states using constrained density functional theory (CDFT). PyCDFT provides an object-oriented, customizable implementation of CDFT, and allows for both single-point self-consistent-field calculations and geometry optimizations. PyCDFT is designed to interface with existing density functional theory (DFT) codes to perform CDFT calculations where constraint potentials are added to the Kohn-Sham Hamiltonian. Here we demonstrate the use of PyCDFT by performing calculations with a massively parallel first-principles molecular dynamics code, Qbox, and we benchmark its accuracy by computing the electronic coupling between diabatic states for a set of organic molecules. We show that PyCDFT yields results in agreement with existing implementations and is a robust and flexible package for performing CDFT calculations. The program is available at https://dx.doi.org/10.5281/zenodo.3821097.
Structure and dynamics of hydrodynamically interacting finite-size Brownian particles in a spherical cavity: Spheres and cylinders
Jiyuan Li, Xikai Jiang, Abhinendra Singh, Olle G. Heinonen, Juan P. Hernández-Ortiz, and Juan J. de Pablo
J. Chem. Phys. 152, 204109 (2020). DOI:10.1063/1.5139431
The structure and dynamics of confined suspensions of particles of arbitrary shape are of interest in multiple disciplines from biology to engineering. Theoretical studies are often limited by the complexity of long-range particle–particle and particle–wall forces, including many-body fluctuating hydrodynamic interactions. Here, we report a computational study on the diffusion of spherical and cylindrical particles confined in a spherical cavity. We rely on an immersed-boundary general geometry Ewald-like method to capture lubrication and long-range hydrodynamics and include appropriate non-slip conditions at the confining walls. A Chebyshev polynomial approximation is used to satisfy the fluctuation–dissipation theorem for the Brownian suspension. We explore how lubrication, long-range hydrodynamics, particle volume fraction, and shape affect the equilibrium structure and the diffusion of the particles. It is found that once the particle volume fraction is greater than 10%, the particles start to form layered aggregates that greatly influence particle dynamics. Hydrodynamic interactions strongly influence the particle diffusion by inducing spatially dependent short-time diffusion coefficients, stronger wall effects on the particle diffusion toward the walls, and a sub-diffusive regime—caused by crowding—in the long-time particle mobility. The level of asymmetry of the cylindrical particles considered here is enough to induce an orientational order in the layered structure, decreasing the diffusion rate and facilitating a transition to the crowded mobility regime at low particle concentrations. Our results offer fundamental insights into the diffusion and distribution of globular and fibrillar proteins inside cells.
Atomistic simulations of the thermal conductivity of liquids
Marcello Puligheddu and Giulia Galli
Phys. Rev. Mat. 4, 053801 (2020). DOI:10.1103/PhysRevMaterials.4.053801
We present a method based on sinusoidal approach to equilibrium molecular dynamics (SAEMD) to compute the thermal conductivity of liquids. Similar to nonequilibrium molecular dynamics, and unlike equilibrium simulations based on the Green-Kubo formalism, the method only requires the calculation of forces and total energies. The evaluation of heat fluxes and energy densities is not necessary, thus offering the promise of efficiently implementing first principles simulations based on density functional theory or deep molecular dynamics. Our approach is a generalization of SAEMD for solids, where the thermal conductivity is computed in the steady state, instead of a transient regime, thus properly taking into account diffusive terms in the heat equation. We present results for liquid water at ambient conditions and under pressure and discuss simulation requirements to obtain converged values of the thermal conductivity as a function of size and simulation time.
Shape induced segregation and anomalous particle transport under spherical confinement
Abhinendra Singh, Jiyuan Li, Xikai Jiang, Juan P. Hernández-Ortiz, Henrich M Jaeger, and Juan J. de Pablo
Phys. Fluids 32, 053307 (2020). DOI:10.1063/5.0002906
Colloidal or nanoparticle mobility under confinement is of central importance for a wide range of physical and biological processes. Here, we introduce a minimal model of particles in a hydrodynamic continuum to examine how particle shape and concentration affect the transport of particles in spherical confinement. Specifically, an immersed boundary-general geometry Ewald-like approach is adopted to simulate the dynamics of spheres and cylinders under the influence of short- and long-range fluctuating hydrodynamic interactions with appropriate non-slip conditions at the confining walls. An efficient O(N) parallel finite element algorithm is used, thereby allowing simulations at high concentrations, while a Chebyshev polynomial approximation is implemented in order to satisfy the fluctuation–dissipation theorem. A concentration-dependent anomalous diffusion is observed for suspended particles. It is found that introducing cylinders in a background of spheres, i.e., particles with a simple degree of anisotropy, has a pronounced influence on the structure and dynamics of the particles. First, increasing the fraction of cylinders induces a particle segregation effect, where spheres are pushed toward the wall and cylinders remain near the center of the cavity. This segregation leads to a lower mobility for the spheres relative to that encountered in a system of pure spheres at the same volume fraction. Second, the diffusive-to-anomalous transition and the degree of anomaly quantified by the power law exponent in the mean square displacement vs time relation both increase as the fraction of cylinders becomes larger. These findings are of relevance for studies of diffusion in the cytoplasm, where proteins exhibit a distribution of size and shapes that could lead to some of the effects identified in the simulations reported here.
Surveying the free energy landscape of clusters of attractive colloidal spheres
Shanghui Huang, Michael J. Quevillon, Soren Kyhl, and Jonathan K. Whitmer
J. Chem. Phys. 152, 134901 (2020). DOI:10.1063/1.5144984
Controlling the assembly of colloidal particles into specific structures has been a long-term goal of the soft materials community. Much can be learned about the process of self-assembly by examining the early stage assembly into clusters. For the simple case of hard spheres with short-range attractions, the rigid clusters of N particles (where N is small) have been enumerated theoretically and tested experimentally. Less is known, however, about how the free energy landscapes are altered when the inter-particle potential is long-ranged. In this work, we demonstrate how adaptive biasing in molecular simulations may be used to pinpoint shifts in the stability of colloidal clusters as the inter-particle potential is varied. We also discuss the generality of our techniques and strategies for application to related molecular systems.
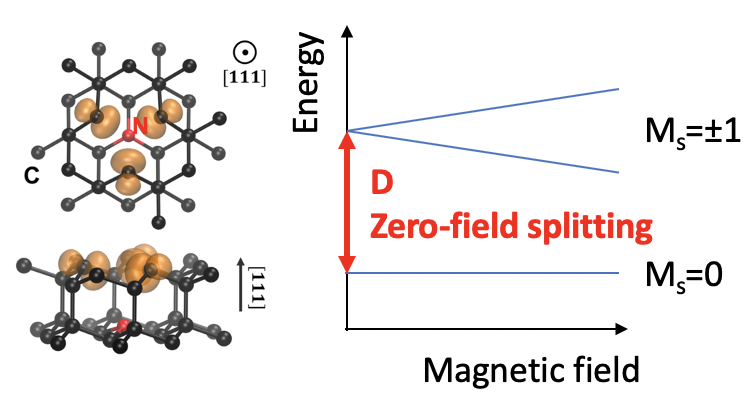
PyZFS: A Python package for first-principles calculations of zero-field splitting tensors
He Ma, Marco Govoni, and Giulia Galli
J. Open Source Softw. 5(47), 2160 (2020). DOI:10.21105/joss.02160
In this work we describe the code PyZFS for the calculation of the ZFS tensor D of molecules and solids, based on wavefunctions obtained from density functional theory (DFT) calculations.
The Long and Winding Road: Predicting Materials Properties Through Theory and Computation
Giulia Galli
First-principles simulations of materials provide both computational microscopes and predictive tools, which we aspire to turn into design strategies for materials with target properties. One requisite to meet this goal is the enablement of predictions of material properties on a large scale, so as to generate a vast amount of validated computational data that may eventually be used to solve inverse problems. However it is challenging to use big data to address the “why question.” First-principles calculations of specific materials and properties can instead be extremely effective at answering the “why question,” namely, at unraveling mechanisms and providing fundamental, physical insights, thus paving the way to innovative design strategies. In this chapter, we present first-principles predictions of material properties relevant to energy conversion processes. We also discuss some open challenges related to automated integration of theory and computation with experiments and with validated, interpreted data.
Machine learning for collective variable discovery and enhanced sampling in biomolecular simulation
Hythem Sidky, Wei Chen, and Andrew L. Ferguson
Mol. Phys. 118, 1-22 (2020). DOI: 10.1080/00268976.2020.1737742
Classical molecular dynamics simulates the time evolution of molecular systems through the phase space spanned by the positions and velocities of the constituent atoms. Molecular-level thermodynamic, kinetic, and structural data extracted from the resulting trajectories provide valuable information for the understanding, engineering, and design of biological and molecular materials. The cost of simulating many-body atomic systems makes simulations of large molecules prohibitively expensive, and the high-dimensionality of the resulting trajectories presents a challenge for analysis. Driven by advances in algorithms, hardware, and data availability, there has been a flare of interest in recent years in the applications of machine learning – especially deep learning – to molecular simulation. These techniques have demonstrated great power and flexibility in both extracting mechanistic understanding of the important nonlinear collective variables governing the dynamics of a molecular system, and in furnishing good low-dimensional system representations with which to perform enhanced sampling or develop long-timescale dynamical models. It is the purpose of this article to introduce the key machine learning approaches, describe how they are married with statistical mechanical theory into domain-specific tools, and detail applications of these approaches in understanding and accelerating biomolecular simulation.
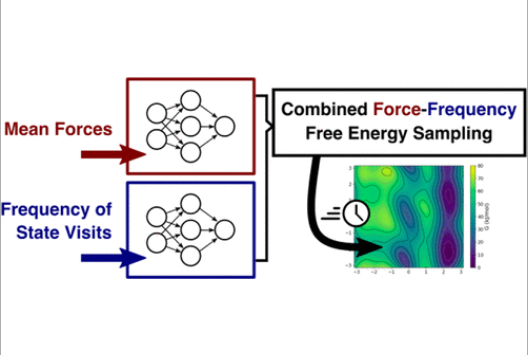
Combined Force-Frequency Sampling for Simulation of Systems Having Rugged Free Energy Landscapes
Emre Sevgen, Ashley Z. Guo, Hythem Sidky, Jonathan K. Whitmer, and Juan J. de Pablo
J. Chem. Theory Comput. 16, 1448-1455 (2020). DOI: 10.1021/acs.jctc.9b00883
An adaptive, machine learning-based sampling method is presented for simulation of systems having rugged, multidimensional free energy landscapes. The method’s main strength resides in its ability to learn both from the frequency of visits to distinct states and the generalized force estimates that arise in a system as it evolves in phase space. This is accomplished by introducing a self-integrating artificial neural network, which generates an estimate of the free energy directly from its derivatives. The usefulness of the proposed combined approach is examined in the context of two concrete examples, namely, an alanine dipeptide molecule in water and a polymer diffusing through a narrow pore. This new method is found to be robust, faster, and more accurate than approaches that rely only on frequency-based or generalized force-based estimations. After combining the proposed approach with overfill protection and support for sparse data storage and training, the method is shown to be more effective than comparable, previously available techniques and capable of scaling efficiently to larger numbers of collective variables.

MatD3: A Database and Online Presentation Package for Research Data Supporting Materials Discovery, Design, and Dissemination
Raul Laasner, Xiaochen Du, Aditya Tanikanti, Connor Clayton, Marco Govoni, Giulia Galli, Matti Ropo, and Volker Blum
J. Open Source Softw. 5(45), 1945 (2020). DOI: 10.21105/joss.01945
MatD3 is an open-source, dedicated database and web application framework designed tostore, curate and disseminate experimental and theoretical materials data generated by indi-vidual research groups or research consortia. A research group can set up its own instance ofMatD3and publish scientific results or simply use an existing online MatD3instance. Dissem-inating research data in this form enables broader access, reproducibility, and repurposing ofscientific products. MatD3is a general purpose database that does not focus on any specificlevel of theory or experimental method. Instead, the focus is on storing and making accessiblethe data and making it straightforward to curate them.
Efficient Multiscale Optoelectronic Prediction for Conjugated Polymers
Nicholas E. Jackson, Alec S. Bowen, and Juan J. de Pablo
Macromolecules 53, 482-490 (2020). DOI: 10.1021/acs.macromol.9b02020
Conjugated polymers represent a high-potential material class due to the tunability of their optoelectronic properties via straightforward processing protocols. However, to in silico tailor these optoelectronic properties, one must compute the conformationally dependent electronic structure over mesoscopic length and time scales. This task represents a challenging multiscale computational problem in which quantum chemistry and atomistic or coarse-grained molecular dynamics must be integrated. Recently, we introduced a methodology, called electronic coarse graining (ECG), that utilizes artificial neural networks to compute ab initio quality electronic structures using only the reduced degrees of freedom of coarse-grained models. Here, we adapt ECG to variable-molecular weight conjugated oligomers by casting the problem in the framework of sequence detection. A machine-learning (ML) architecture employing one-dimensional convolutional neural networks and long short-term memory networks is utilized to compute ground-state orbital energies, charge density distributions, and optical spectra solely from the coarse-grained model’s configurational degrees of freedom. Robust molecular weight transferability for ECG is established via a Δ-ML approach that leverages model electronic Hamiltonians for ground and excited-state property determination. The accuracy and transferability of the ECG methodology opens the door for scalable optoelectronic property prediction in conjugated polymers directly from coarse-grained degrees of freedom.
Improving the efficiency of G0W0 calculations with approximate spectral decompositions of dielectric matrices
Han Yang, Marco Govoni, and Giulia Galli
J. Chem. Phys. 151, 224102 (2019). DOI: 10.1063/1.5126214
Recently it was shown that the calculation of quasiparticle energies using the G0W0 approximation can be performed without computing explicitly any virtual electronic states, by expanding the Green function and screened Coulomb interaction in terms of the eigenstates of the static dielectric matrix. Avoiding the evaluation of virtual electronic states leads to improved efficiency and ease: of convergence of G0W0 calculations. Here we propose a further improvement of the efficiency of these calculations, based on an approximation of density-density response functions of molecules and solids. The approximation relies on the calculation of a subset of eigenvectors of the dielectric matrix using the kinetic operator instead of the full Hamiltonian, and it does not lead to any substantial loss of accuracy for the quasiparticle energies. The computational savings introduced by this approximation depend on the system, and they become more substantial as the number of electrons increases.
Influence of Softness on the Stability of Binary Colloidal Crystals
R. Allen LaCour, Carl Simon Adorf, Julia Dshemuchadse, Sharon C. Glotzer
ACS Nano 15, 5761-5768 (2019). DOI: 10.1021/acsnano.9b04274
Mixtures of two types of nanoparticles can self-assemble into a wide variety of binary colloidal crystals (also called binary nanoparticle superlattices), which are interesting for their structural diversity and potential applications. Although so-called packing models—which usually treat the particles as “hard” with only excluded volume interactions—seem to explain many reported dense crystalline phases, these models often fail to predict the right structure. Here, we examine the role of soft repulsive interparticle interactions on binary colloidal crystals comprising two sizes of spherical particles; such “softness” can arise due to ligand shells or screened electrostatics. We determine the ground state phase diagram of binary systems of particles interacting with an additive inverse power law potential using a basin hopping algorithm to calculate the enthalpy of an extremely large pool of candidate structures. We find that a surprisingly small amount of softness can destabilize dense packings in favor of less densely packed structures, which provides further evidence that considerations beyond packing are necessary for describing many of the observed phases of binary colloidal crystals. Importantly, we find that several of the phases stabilized by softness, which are characterized by relatively few interparticle contacts and a tendency for local icosahedral order, are more likely to be observed experimentally than those predicted by packing models. We also report a previously unknown dense AB4 phase and conduct free energy calculations to examine how the stability of several crystals will vary with temperature. Our results further our understanding of why particular binary colloidal crystals form and will be useful as a reference for experimentalists working with softly repulsive colloids.
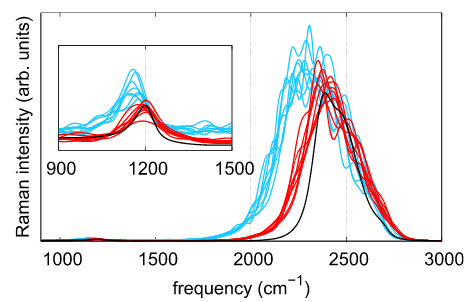
Ensemble first-principles molecular dynamics simulations of water using the SCAN meta-GGA density functional
Michael D. LaCount and Francois Gygi
J. Chem. Phys. 151, 164101 (2019), Editor's Pick. DOI: 10.1063/1.5124957
We present an ensemble of 16 independent first-principles molecular dynamics simulations of water performed using the Strongly Constrained and Appropriately Normed (SCAN) meta-generalized gradient approximation exchange-correlation functional. These simulations were used to compute the structural and electronic properties of liquid water, as well as polarizabilities, Raman and infrared spectra. Overall, we find that the SCAN functional used at a simulation temperature of 330 K provides an accurate description of the structural and electronic properties of water while incurring a moderate computational cost. The availability of an ensemble of independent simulations provides a quantitative estimate of the uncertainty in computed structural and electronic properties. Results are also compared with a similar dataset generated using the Perdew, Burke, and Ernzerhof exchange-correlation functional at a temperature of 400 K. All simulation data and trajectories are available at http://quantum-simulation.org.
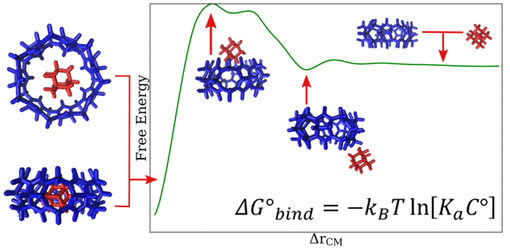
Accurate Determination of Cavitand Binding Free Energies via Unrestrained Advanced Sampling
Anne C. Leonhard and Jonathan K. Whitmer
J. Chem. Theory Comput. 15, 5761-5768 (2019). DOI: 10.1021/acs.jctc.9b00348
Calculation of binding free energies, which characterize the stability and specificity of molecular interactions (e.g., drug formulations), has been established as an important application of modern computational chemistry. Though several methodologies currently exist within the literature, there is always a need for improvements in speed and accuracy that allow for efficient and accurate chemical screening. In this paper we explore the application of an unrestrained advanced sampling method, adaptive biasing force (ABF), to the calculation of standard binding free energies for a set of molecular host–guest systems for which the binding free energy is well-known: the cucurbit[7]uril-hosted systems utilized in the HYDROPHOBE challenge [Assaf et al. J. Phys. Chem. B 2017, 121, 11144−11162]. We demonstrate that the use of the ABF advanced sampling method yields values systematically closer to experiment, with a lower uncertainty in the calculation, than thermodynamic integration and restrained sampling techniques commonly used in the literature. We hypothesize that this is due to the algorithm’s ability to drive multiple binding and dissociation events through applied bias forces. These results show the promise of unrestrained advanced sampling methods along well-defined reaction coordinates to the calculation of binding energies in host–guest systems.
Novel elastic response in twist-bend nematic models
Jiale Shi, Hythem Sidky, and Jonathan K. Whitmer
Soft Matter 15, 8219-8226 (2019). DOI: 10.1039/C9SM01395D
Bent-shaped liquid crystals have attracted significant attention recently due to their novel mesostructure and the intriguing behavior of their elastic constants, which are strongly anisotropic and have an unusual temperature dependence. Though theories explain the onset of the twist-bend nematic phase (NTB) through spontaneous symmetry breaking concomitant with transition to a negative bend (K3) elastic constant, this has not been observed as yet in experiments. There, the small bend elastic constant has a strongly non-monotonic temperature dependence, which first increases after crossing the isotropic (I)–nematic (N) transition, then dips near the nematic (N)-twist-bend (NTB) transition before it increases again as the transition is crossed. The molecular mechanisms responsible for this exotic behavior are unclear. Here, we utilize density of states algorithms in Monte Carlo simulation applied to a variant of the Lebwohl–Lasher model which includes bent-shaped-like interactions to analyze the mechanism behind elastic response in this novel mesostructure and understand the temperature dependence of its Frank–Oseen elastic constants.
Colloidal Atomic Layer Deposition with Stationary Reactant Phases Enables Precise Synthesis of “Digital” II–VI Nano-heterostructures with Exquisite Control of Confinement and Strain
Abhijit Hazarika, Igor Fedin, Liang Hong, Jinglong Guo, Vishwas Srivastava, Wooje Cho, Igor Coropceanu, Joshua Portner, Benjamin T. Diroll, John P. Philbin, Eran Rabani, Robert Klie, and Dmitri V. Talapin
J. Amer. Chem. Soc. 141, 13487-13496 (2019). DOI: 10.1021/jacs.9b04866
In contrast to molecular systems, which are defined with atomic precision, nanomaterials generally show some heterogeneity in size, shape, and composition. The sample inhomogeneity translates into a distribution of energy levels, band gaps, work functions, and other characteristics, which detrimentally affect practically every property of functional nanomaterials. We discuss a novel synthetic strategy, colloidal atomic layer deposition (c-ALD) with stationary reactant phases, which largely circumvents the limitations of traditional colloidal syntheses of nano-heterostructures with atomic precision. This approach allows for significant reduction of inhomogeneity in nanomaterials in complex nanostructures without compromising their structural perfection and enables the synthesis of epitaxial nano-heterostructures of unprecedented complexity. The improved synthetic control ultimately enables bandgap and strain engineering in colloidal nanomaterials with close to atomic accuracy. To demonstrate the power of the new c-ALD method, we synthesize a library of complex II–VI semiconductor nanoplatelet heterostructures. By combining spectroscopic and computational studies, we elucidate the subtle interplay between quantum confinement and strain effects on the optical properties of semiconductor nanostructures.
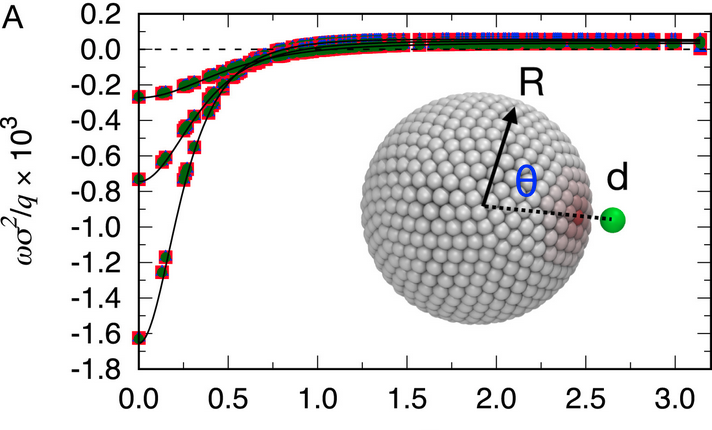
Incorporating surface polarization effects into large-scale coarse-grained Molecular Dynamics simulation
Trung Dac Nguyen, Honghao Li, Debarshee Bagchi, Francisco J. Solis, and Monica Olvera de la Cruz
Comp. Phys. Commun. 241, 80-91 (2019). DOI: 10.1016/j.cpc.2019.03.006
We implement and extend three methods for incorporating surface polarization effects into coarse-grained Molecular Dynamics (MD) simulations for LAMMPS: the boundary element method using the generalized minimum residual (GMRES) solver for solving the Poisson equation, the induced charge computation method using the successive over relaxations scheme, and the direct optimization of an energy functional of induced charge density. Our implementations are validated against the analytical results for several cases and against published results on electrolyte and polyelectrolyte adsorption on charged interfaces. We have also employed the mentioned methods to examine the effects of surface polarization due to dielectric mismatch on the conformational behavior and relaxation times of the Rouse modes of a highly charged polyelectrolyte with explicit multivalent counterions in a spherical droplet surrounded by a low dielectric medium. We have found that dielectric mismatch coupled with spatial confinement results in noticeable changes in the radius of gyration of the collapsed conformation as well as the work required to compress and stretch the chain. Dielectric confinement is therefore believed to play an important role in the packaging of polyelectrolytes, which was generally overlooked in previous studies. Finally, the parallel performance of the implemented methods for practical simulations is characterized for a model electrolyte system. For polarizable interfaces composed of more than 1000 mesh points, the induced charge computation method is found to be the most efficient.

Emergence of Radial Tree of Bend Stripes in Active Nematics
Andrey Sokolov, Ali Mozaffari, Rui Zhang, Juan J. de Pablo, and Alexey Snezhko
Phys. Rev. X 9, 031014 (2019). DOI: 10.1103/PhysRevX.9.031014
Living liquid crystals, a realization of active nematics where a lyotropic liquid crystal is combined with active bacteria, exhibit a plethora of out-of-equilibrium phenomena that range from active turbulence and dynamic spatiotemporal patterns to the creation and annihilation of motile topological defects. Experiments and hydrodynamic simulations are used here to report on the emergence of bend stripes, which arise as spontaneous undulations of the director field in circularly aligned lyotropic liquid crystals doped with bacteria. The interplay between bacterial-induced hydrodynamic flows and elastic forces in the material induces remarkable deformation patterns consisting of branched, radially elongated bands of a high curvature of the director field. The average number of such branches increases with the distance from the center of the circular alignment, leading to the formation of a radial tree of bands that is reminiscent of a snowflake structure. Hydrodynamic simulations, which are in agreement with the experiments, are used to explain the origin of such structures and to provide additional insights into regimes that are beyond the limit of experimental measurements. In particular, it is found that when activity is switched off in the early stages of pattern formation, a pronounced decay of bend-distortion energy ensues, with little change of the splay energy, serving to confirm that the bend stripes are an outcome of activity-driven bend-instability phenomena. Taken together, experiments and simulations demonstrate a system in which strain and geometry can be combined to dynamically manipulate pattern formation in active matter, paving the way to a deeper understanding and finer control of active colloidal systems.
Dielectric dependent hybrid functionals for heterogeneous materials
Huihuo Zheng, Marco Govoni, and Giulia Galli
Phys. Rev. Mat. 3, 073803 (2019). DOI: 10.1103/PhysRevMaterials.3.073803
We derive a dielectric-dependent hybrid functional which accurately describes the electronic properties of heterogeneous interfaces and surfaces, as well as those of three- and two-dimensional bulk solids. The functional, which does not contain any adjustable parameter, is a generalization of self-consistent hybrid functionals introduced for homogeneous solids, where the screened Coulomb interaction is defined using a spatially varying, local dielectric function. The latter is determined self-consistently using density functional calculations in finite electric fields. We present results for the band gaps and dielectric constants of 3D and 2D bulk materials, and band offsets for interfaces, showing an accuracy comparable to that of GW calculations.
Computational prediction of lattice thermal conductivity -- a comparison of molecular dynamics and Boltzmann transport approaches
Marcello Puligheddu, Yi Xia, Maria K. Y. Chan, and Giulia Galli
Phys. Rev. Mat. 3, 085401 (2019). DOI: 10.1103/PhysRevMaterials.3.085401.
The predictive modeling of lattice thermal conductivity is of fundamental importance for the understanding and design of materials for a wide range of applications. Two major approaches, namely molecular dynamics (MD) simulations and calculations solving approximately the Boltzmann transport equation (BTE), have been developed to compute the lattice thermal conductivity. We present a detailed direct comparison of these two approaches, using as prototypical cases MgO and PbTe. The comparison, carried out using empirical potentials, takes into account the effects of fourth order phonon scattering, temperature-dependent phonon frequencies (phonon renormalization), and investigates the effects of quantum vs. classical statistics. We clarify that equipartition, as opposed to Maxwell Boltzmann, govern the statistics of phonons in MD simulations. We find that lattice thermal conductivity values from MD and BTE show an apparent, satisfactory agreement; however such an agreement is the result of error cancellations. We also show that the primary effect of statistics on thermal conductivity is via the scattering rate dependence on phonon populations.
1CPN: A coarse-grained multi-scale model of chromatin
Joshua Lequieu, Andrés Córdoba, Joshua Moller, and Juan J. de Pablo
J. Chem. Phys. 150, 215102 (2019). DOI: 10.1063/1.5092976
A central question in epigenetics is how histone modifications influence the 3D structure of eukaryotic genomes and, ultimately, how this 3D structure is manifested in gene expression. The wide range of length scales that influence the 3D genome structure presents important challenges; epigenetic modifications to histones occur on scales of angstroms, yet the resulting effects of these modifications on genome structure can span micrometers. There is a scarcity of computational tools capable of providing a mechanistic picture of how molecular information from individual histones is propagated up to large regions of the genome. In this work, a new molecular model of chromatin is presented that provides such a picture. This new model, referred to as 1CPN, is structured around a rigorous multiscale approach, whereby free energies from an established and extensively validated model of the nucleosome are mapped onto a reduced coarse-grained topology. As such, 1CPN incorporates detailed physics from the nucleosome, such as histone modifications and DNA sequence, while maintaining the computational efficiency that is required to permit kilobase-scale simulations of genomic DNA. The 1CPN model reproduces the free energies and dynamics of both single nucleosomes and short chromatin fibers, and it is shown to be compatible with recently developed models of the linker histone. It is applied here to examine the effects of the linker DNA on the free energies of chromatin assembly and to demonstrate that these free energies are strongly dependent on the linker DNA length, pitch, and even DNA sequence. The 1CPN model is implemented in the LAMMPS simulation package and is distributed freely for public use.
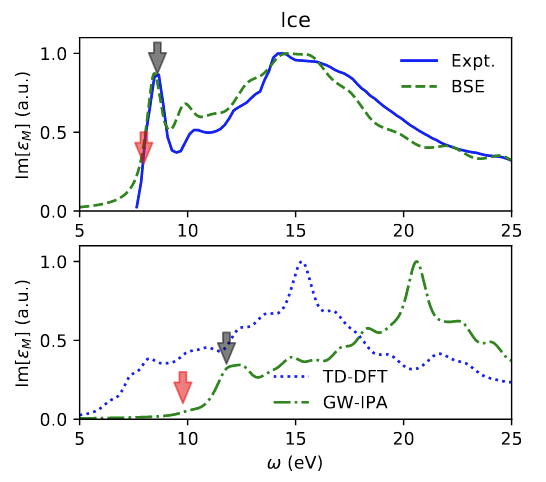
Finite field approach to solving the Bethe Salpeter equation
Ngoc Linh Nguyen, He Ma, Marco Govoni, Francois Gygi, and Giulia Galli
Phys. Rev. Lett. 122, 237402 (2019). DOI: 10.1103/PhysRevLett.122.237402
We present a method to compute optical spectra and exciton binding energies of molecules and solids based on the solution of the Bethe-Salpeter equation (BSE) and the calculation of the screened Coulomb interaction in finite field. The method does not require the explicit evaluation of dielectric matrices nor of virtual electronic states, and can be easily applied without resorting to the random phase approximation. In addition it utilizes localized orbitals obtained from Bloch states using bisection techniques, thus greatly reducing the complexity of the calculation and enabling the efficient use of hybrid functionals to obtain single particle wavefunctions. We report exciton binding energies of several molecules and absorption spectra of condensed systems of unprecedented size, including water and ice samples with hundreds of atoms.
Structure and proton conduction in sulfonated poly(ether ether ketone) semi-permeable membranes: a multi-scale computational approach
Jarol Molina, Juan J. de Pablo, and Juan P. Hernández-Ortiz
Phys. Chem. Chem. Phys. 21, 9362 (2019). DOI: 10.1039/c9cp00598f
The design of polymeric membranes for proton or ionic exchange highly depends on the fundamentalunderstanding of the physical and molecular mechanisms that control the formation of the conductionchannels. There is an inherent relation between the dynamical structure of the polymeric membraneand the electrostatic forces that drive membrane segregation and proton transport. Here, we used amulti-scale computational approach to analyze the morphology of sulfonated poly(ether ether ketone)membranes at the mesoscale. A self-consistent description of the electrostatic phenomenon wasadopted, where discrete polymer chains and a continuum proton field were embedded in a continuumfluid. Brownian dynamics was used for the evolution of the suspended polymer molecules, while aconvection–diffusion transport equation, including the Nernst–Planck diffusion mechanism, accountedfor the dynamics of the proton concentration field. We varied the polymer concentration, the degree ofsulfonation and the level of confinement to find relationships between membrane structure and protonconduction. Our results indicate that the reduced mobility of polymer chains, at concentrations aboveoverlap, and a moderate degree of sulfonation –i.e., 30% – are essential elements for membrane segregationand proton domain connectivity. These conditions also ensure that the membrane structure is not affectedby size or by potential gradients. Importantly, our analysis shows that membrane conductivity and current arelinearly dependent on polymer concentration and quadratically dependent on the degree of sulfonation. Wefound that the optimal polymeric membrane design requires a polymer concentration above overlap and adegree of sulfonation around 50%. These conditions promote a dynamical membrane morphology with aconstant density of proton channels. Our results and measurements agree with previous experimental works,thereby validating our model and observations.
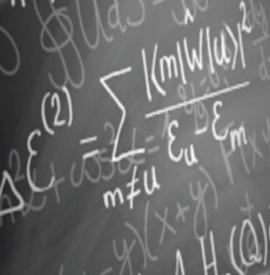
All-electron density functional calculations for electron and nuclear spin interactions in molecules and solids
Krishnendu Ghosh, He Ma, Vikram Gavini, and Giulia Galli
Phys. Rev. Mat. 3, 043801 (2019). DOI: 10.1103/PhysRevMaterials.3.043801
The interaction between electronic and nuclear spins in the presence of external magnetic fields can be described by a spin Hamiltonian, with parameters obtained from first principles, electronic structure calculations. We describe an approach to compute these parameters, applicable to both molecules and solids, which is based on Density Functional Theory (DFT) and real-space, all-electron calculations using finite elements (FE). We report results for hyperfine tensors, zero field splitting tensors (spin-spin component) and nuclear quadrupole tensors of a series of molecules and of the nitrogen-vacancy center in diamond. We compare our results with those of calculations using Gaussian orbitals and plane-wave basis sets, and we discuss their numerical accuracy. We show that calculations based on FE can be systematically converged with respect to the basis set, thus allowing one to establish reference values for the spin Hamiltonian parameters, at a given level of DFT.
Recent advances in machine learning towards multiscale soft materials design
Nicholas E. Jackson, Michael A. Webb, and Juan J. de Pablo
Curr. Opin. Chem. Eng. 23, 106-114, (2019). DOI: 10.1016/j.coche.2019.03.005
The multiscale design of soft materials requires an ensemble of computational techniques spanning quantum-chemistry to molecular dynamics to continuum modeling. The recent emergence of machine-learning (ML) and modern optimization algorithms has accelerated material property prediction, as well as stimulated the development of hybrid ML/molecular modeling methodologies capable of providing physical insights unobtainable from purely physics-based modeling and intuition. Such hybrid techniques also have important ramifications for the ML-enhanced interpretation of results from simulations and experiments alike. Leveraging ML techniques for the design of chemical or morphological structures based on a target property or functionality represents an exciting goal for the general area of soft materials, including polymers, liquid crystals, colloids, or biomolecules, to name a few representative classes of systems. Here, we provide a perspective on recent work using ML techniques of relevance for the multiscale design of soft materials and outline potential future directions of interest to the soft materials community.
Degenerate conic anchoring and colloidal elastic dipole-hexadecapole transformations
Ye Zhou, Bohdan Senyuk, Rui Zhang, Ivan I. Smalyukh, and Juan J. de Pablo
Nat. Commun. 10, 1000, (2019). DOI: 10.1038/s41467-019-08645-9
The defect structure associated with a colloid in a nematic liquid crystal is dictated bymolecular orientation at the colloid surface. Perpendicular or parallel orientations to thesurface lead to dipole-like or quadrupole-like defect structures. However, the so-called elastichexadecapole discovered recently, has been assumed to result from a conic anchoringcondition. In order to understand it at a fundamental level, a model for this anchoring isintroduced here in the context of a Landau-de Gennes free energy functional. We investigatethe evolution of defect configurations, as well as colloidal interactions, by tuning the preferredtilt angle (θe). The model predicts an elastic dipole whose stability decreases asθeincreases,along with a dipole-hexadecapole transformation, which are confirmed by our experimentalobservations. Taken together, our results suggest that previously unanticipated avenues mayexist for design of self-assembled structures via control of tilt angle.
Systematic Mapping of Binary Nanocrystal Superlattices: The Role of Topology in Phase Selection
Igor Coropceanu, Michael A. Boles, and Dmitri V. Talapin
J. Amer. Chem. Soc. 141, 5728 (2019). DOI: 10.1021/jacs.8b12539
The self-assembly of two sizes of spherical nanocrystals has revealed a surprisingly diverse library of structures. To date, at least 15 distinct binary nanocrystal superlattice (BNSL) structures have been identified. The stability of these binary phases cannot be fully explained using the traditional conceptual framework treating the assembly process as entropy-driven crystallization of rigid spherical particles. Such deviation from hard sphere behavior may be explained by the soft and deformable layer of ligands that envelops the nanocrystals, which contributes significantly to the overall size and shape of assembling particles. In this work, we describe a set of experiments designed to elucidate the role of the ligand corona in shaping the thermodynamics and kinetics of BNSL assembly. Using hydrocarbon-capped Au and PbS nanocrystals as a model binary system, we systematically tuned the core radius (R) and ligand chain length (L) of particles and subsequently assembled them into binary superlattices. The resulting database of binary structures enabled a detailed analysis of the role of effective nanocrystal size ratio, as well as softness expressed as L/R, in directing the assembly of binary structures. This catalog of superlattices allowed us to not only study the frequency of different phases but to also systematically measure the geometric parameters of the BNSLs. This analysis allowed us to evaluate new theoretical models treating the cocrystallization of deformable spheres and to formulate new hypotheses about the factors affecting the nucleation and growth of the binary superlattices. Among other insights, our results suggest that the relative abundance of the binary phases observed may be explained not only by considerations of thermodynamic stability, but also by a postulated preordering of the binary fluid into local structures with icosahedral or polytetrahedral symmetry prior to nucleation.
Electronic structure at coarse-grained resolutions from supervised machine learning
Nicholas E. Jackson, Alec S. Bowen, Lucas W. Antony, Michael A. Webb, Venkatram Vishwanath, and Juan J. de Pablo
Sci. Adv. 5, eaav1190 (2019). DOI: 10.1126/sciadv.aav1190
Computational studies aimed at understanding conformationally dependent electronic structure in soft materials require a combination of classical and quantum-mechanical simulations, for which the sampling of conformational space can be particularly demanding. Coarse-grained (CG) models provide a means of accessing relevant time scales, but CG configurations must be back-mapped into atomistic representations to perform quantum-chemical calculations, which is computationally intensive and inconsistent with the spatial resolution of the CG models. A machine learning approach, denoted as artificial neural network electronic coarse graining (ANN-ECG), is presented here in which the conformationally dependent electronic structure of a molecule is mapped directly to CG pseudo-atom configurations. By averaging over decimated degrees of freedom, ANN-ECG accelerates simulations by eliminating backmapping and repeated quantum-chemical calculations. The approach is accurate, consistent with the CG spatial resolution, and can be used to identify computationally optimal CG resolutions.
Free energy of metal-organic framework self-assembly
Yamil J. Colon, Ashley Z. Guo, Lucas W. Antony, Kyle Q. Hoffmann, and Juan J. de Pablo
J. Chem. Phys 150, 104502 (2019). DOI: 10.1063/1.5063588
Metal-organic frameworks (MOFs) represent an important class of materials. Careful selection of building blocks allows for tailoring of the properties of the resulting framework. The self-assembly process, however, is not understood, and without detailed knowledge of the underlying molecular mechanism, it is difficult to anticipate whether a particular design can be realized, or whether the material adopts a metastable, kinetically arrested state. We present a detailed examination of early-stage self-assembly pathways of the MOF-5. Enhanced sampling techniques are used to model a self-assembly in an explicit solvent (dimethylformamide, DMF). We identify several free energy barriers encountered during the assembly of the final MOF, which arise from structural rearrangements preceding MOF formation and from disrupted MOF-solvent interactions as formation proceeds. In all cases considered here, MOFs exhibit favorable entropic gains during the assembly. More generally, the strategy presented provides a step toward the experimental design characterizing the formation of ordered frameworks and possible sources of polymorphism.

Qresp, a Tool for Curating, Discovering and Exploring Reproducible Scientific Papers
Marco Govoni, Milson Munakami, Aditya Tanikanti, Jonathan H. Skone, Hakizumwami B. Runesha, Federico Giberti, Juan J. de Pablo, and Giulia Galli
Sci. Data 6, 190002 (2019). DOI: 10.1038/sdata.2019.2
We propose a strategy and present a simple tool to facilitate scientific data reproducibility by making available, in a distributed manner, all data and procedures presented in scientific papers, together with metadata to render them searchable and discoverable. In particular, we describe a graphical user interface (GUI), Qresp, to curate papers (i.e. generate metadata) and to explore curated papers and automatically access the data presented in scientific publications.
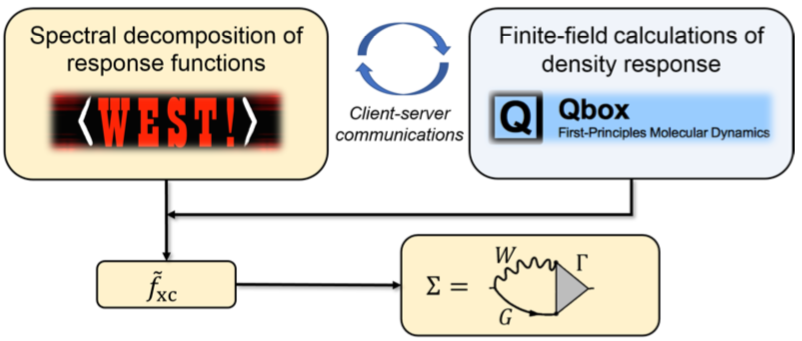
A Finite-field Approach for GW Calculations Beyond the Random Phase Approximation
He Ma, Marco Govoni, Francois Gygi, and Giulia Galli
J. Chem. Theory Comput. 15, 154-164 (2019). DOI: 10.1021/acs.jctc.8b00864
We describe a finite-field approach to compute density response functions, which allows for efficient G0W0 and G0W0Γ0 calculations beyond the random phase approximation. The method is easily applicable to density functional calculations performed with hybrid functionals. We present results for the electronic properties of molecules and solids and we discuss a general scheme to overcome slow convergence of quasiparticle energies obtained from G0W0Γ0 calculations, as a function of the basis set used to represent the dielectric matrix.

Explicit Ion Effects on the Charge and Conformation of Weak Polyelectrolytes
Vikramjit S. Rathee, Hythem Sidky, Benjamin J. Sikora, and Jonathan K. Whitmer
Polymers 11, 183 (2019). DOI: 10.3390/polym11010183
The titration behavior of weak polyelectrolytes is of high importance, due to their uses in new technologies including nanofiltration and drug delivery applications. A comprehensive picture of polyelectrolyte titration under relevant conditions is currently lacking, due to the complexity of systems involved in the process. One must contend with the inherent structural and solvation properties of the polymer, the presence of counterions, and local chemical equilibria enforced by background salt concentration and solution acidity. Moreover, for these cases, the systems of interest have locally high concentrations of monomers, induced by polymer connectivity or confinement, and thus deviate from ideal titration behavior. This work furthers knowledge in this limit utilizing hybrid Monte Carlo–Molecular Dynamics simulations to investigate the influence of salt concentration, pKa, pH, and counterion valence in determining the coil-to-globule transition of poorly solvated weak polyelectrolytes. We characterize this transition at a range of experimentally relevant salt concentrations and explicitly examine the role multivalent salts play in determining polyelectrolyte ionization behavior and conformations. These simulations serve as an essential starting point in understanding the complexation between weak polyelectrolytes and ion rejection of self-assembled copolymer membranes
A Graph-based Approach to Systematic Molecular Coarse-graining
Michael Webb, Jean-Yves Delannoy, and Juan J. de Pablo
J. Chem. Theory Comput. 15, 1199-1208 (2019). DOI: 10.1021/acs.jctc.8b00920
A novel methodology is introduced here to generate coarse-grained (CG) representations of molecular models for simulations. The proposed strategy relies on basic graph-theoretic principles, and is referred to as graph-based coarse-graining (GBCG). It treats a given system as a molecular graph, and derives a corresponding CG representation by using edge contractions to combine nodes in the graph, which correspond to atoms in the molecule, into CG sites. A key element of this methodology is that the nodes are combined according to well defined protocols that rank-order nodes based on the underlying chemical connectivity. By iteratively performing these operations, successively coarser representations of the original atomic system can be produced to yield a systematic set of CG mappings with hierarchical resolution in an automated fashion. These capabilities are demonstrated in the context of several test systems, including toluene, pentadecane, a polysaccharide dimer, and a rhodopsin protein. In these examples, GBCG yields multiple, intuitive structures that naturally preserve the chemical topology of the system. Importantly, these representations are rendered from algorithmic implementation rather than an arbitrary ansatz, which, until now, has been the conventional approach for defining CG mapping schemes. Overall, the results presented here indicate that GBCG is efficient, robust, and unambiguous in its application, making it a valuable tool for future CG modeling.
Fundamental Principles for Calculating Charged Defect Ionization Energies in Ultrathin Two-Dimensional Materials
T.J. Smart, F. Wu, M. Govoni, and Y. Ping
Phys. Rev. Mat. 2, 124002 (2018). DOI: 10.1103/PhysRevMaterials.2.124002
Defects in 2D materials are becoming prominent candidates for quantum emitters and scalable optoelectronic applications. However, several physical properties that characterize their behavior, such as charged defect ionization energies, are difficult to simulate with conventional first-principles methods, mainly because of the weak and anisotropic dielectric screening caused by the reduced dimensionality. We establish fundamental principles for accurate and efficient calculations of charged defect ionization energies and electronic structure in ultrathin 2D materials. We propose to use the vacuum level as the reference for defect charge transition levels (CTLs) because it gives robust results insensitive to the level of theory, unlike commonly used band edge positions. Furthermore, we determine the fraction of Fock exchange in hybrid functionals for accurate band gaps and band edge positions of 2D materials by enforcing the generalized Koopmans’ condition of localized defect states. We found the obtained fractions of Fock exchange vary significantly from 0.2 for bulk h-BN to 0.4 for monolayer h-BN, whose band gaps are also in good agreement with experimental results and calculated GW results. The combination of these methods allows for reliable and efficient prediction of defect ionization energies (difference between CTLs and band edge positions). We motivate and generalize these findings with several examples including different defects in monolayer to few-layer hexagonal boron nitride (h-BN), monolayer MoS2 and graphane. Finally, we show that increasing the number of layers ofh-BN systematically lowers defect ionization energies, mainly through CTLs shifting towards vacuum, with conduction band minima kept almost unchanged.
How to Professionally Develop Reusable Scientific Software—And When Not To
Carl S. Adorf, Vyas Ramasubramani, Joshua A. Anderson, and Sharon C. Glotzer
Comput. Sci. Eng. 21, 66-79 (2018). DOI: 10.1109/mcse.2018.2882355
A critical challenge in scientific computing is balancing developing high quality software with the need for immediate scientific progress. We present a flexible approach that emphasizes writing specialized code that is refactored only when future immediate scientific goals demand it. Our lazy refactoringtechnique, which calls for code with clearly defined interfaces and sharply delimited scopes to maximize reuse and integrability, helps reduce total development time and accelerates the production of scientific results. We offer guidelines for how to implement such code, as well as criteria to aid in the evaluation of existing tools. To demonstrate their application, we showcase the development progression of tools for particle simulations originating from the Glotzer Group at the University of Michigan. We emphasize the evolution of these tools into a loosely integrated software stack of highly reusable software that can be maintained to ensure the long-term stability of established research workflows.
Chemical Insights into PbSe-x%HgSe: High Power Factor and Improved Thermoelectric Performance by Alloying with Discordant Atoms
James M Hodges, Shiqiang Hao, Jann A Grovogui, Xiaomi Zhang, Trevor Paul Bailey, Xiang Li, Zhehong Gan, Yan-Yan Hu, Ctirad Uher, Vinayak P. Dravid, Christopher Wolverton, and Mercouri G. Kanatzidis
J. Am. Chem. Soc. 140, 18115-18123 (2018). DOI: 10.1021/jacs.8b11050
Thermoelectric generators can convert heat directly into usable electric power but suffer from low efficiencies and high costs, which have hindered wide-scale applications. Accordingly, an important goal in the field of thermoelectricity is to develop new high performance materials that are composed of more earth-abundant elements. The best systems for midtemperature power generation rely on heavily doped PbTe, but the Te in these materials is scarce in the Earth’s crust. PbSe is emerging as a less expensive alternative to PbTe, although it displays inferior performance due to a considerably smaller power factor S2σ, where S is the Seebeck coefficient and σ is electrical conductivity. Here, we present a new p-type PbSe system, Pb0.98Na0.02Se–x%HgSe, which yields a very high power factor of ∼20 μW·cm–1·K–2 at 963 K when x = 2, a 15% improvement over the best performing PbSe–x%MSe materials. The enhancement is attributed to a combination of high carrier mobility and the early onset of band convergence in the Hg-alloyed samples (∼550 K), which results in a significant increase in the Seebeck coefficient. Interestingly, we find that the Hg2+ cations sit at an off-centered position within the PbSe lattice, and we dub the displaced Hg atoms “discordant”. DFT calculations indicate that this feature plays a role in lowering thermal conductivity, and we believe that this insight may inspire new design criteria for engineering high performance thermoelectric materials. The high power factor combined with a decrease in thermal conductivity gives a high figure of merit ZT of 1.7 at 970 K, the highest value reported for p-type PbSe to date.
Nanocrystalline Oligo(ethylene sulfide)-b-poly(ethylene glycol) Micelles: Structure and Stability
Emre Sevgen, Moshe Dolejsi, Paul F. Nealey, Jeffrey A. Hubbell, and Juan J. de Pablo
Macromolecules 51, 9538-9546 (2018). DOI: 10.1021/acs.macromol.8b01812
Micelle formation generally relies on hydrophobic or electrostatic interactions between distinct regions of amphiphilic molecules. In this work, a different mechanism is considered in which nanocrystalline domains are formed from short ethylene sulfide oligomers at the core of the micelles, leading to exceptionally stable, uniform micellar structures. The structure and thermodynamic properties of the resulting micelles are examined through a combination of experiments, theory, and simulations. It is found that in oligo(ethylene sulfide)-b-poly(ethylene glycol) amphiphiles as few as three ethylene sulfide monomers are sufficient to form a highly crystalline core, surrounded by a water-soluble ethylene glycol corona of arbitrary size. Sulfur–sulfur interactions induce formation of rhombohedral lattice crystalline regions, which exhibit well-defined intramolecular and intermolecular order. An atomistic model is used to determine the free energy of the micelles; the critical micelle concentration (CMC) is found to be extremely small, on the order of 10–8 mol/L. The size distribution of these micelles is nearly monodisperse. The crystalline core also includes amorphous regions that could serve as hosts for other molecules. Taken together, these properties serve to underscore that controlled crystallization provides a useful and underexploited mechanism for assembly of ultrastable micelles for applications in a variety of settings, including drug delivery and immunology.
Role of Associative Charging in the Entropy–Energy Balance of Polyelectrolyte Complexes
Vikramjit S. Rathee, Hythem Sidky, Benjamin J. Sikora, and Jonathan K. Whitmer
J. Am. Chem. Soc. 140, 15319–15328 (2018). DOI: 10.1021/jacs.8b08649
Polyelectrolytes may be classified into two primary categories (strong and weak) depending on how their charge state responds to the local environment. Both of these find use in many applications, including drug delivery, gene therapy, layer-by-layer films, and fabrication of ion filtration membranes. The mechanism of polyelectrolyte complexation is, however, still not completely understood, though experimental investigations suggest that entropy gain due to release of counterions is the key driving force for strong polyelectrolyte complexation. Here we perform a comprehensive thermodynamic investigation through coarse-grained molecular simulations permitting us to calculate the free energy of complex formation. Importantly, our expanded-ensemble methods permit the explicit separation of energetic and entropic contributions to the free energy. Our investigations indicate that entropic contributions indeed dominate the free energy of complex formation for strong polyelectrolytes, but are less important than energetic contributions when weak electrostatic coupling or weak polyelectrolytes are present. Our results provide a new view of the free energy of polyelectrolyte complex formation driven by polymer association, which should also arise in systems with large charge spacings or bulky counterions, both of which act to weaken ion–polymer binding.
Coupling First Principles Calculations of Electron-Electron and Electron-Phonon Scattering, and Applications to Carbon-based Nanostructures
Ryan McAvoy, Marco Govoni, and Giulia Galli
J. Chem. Theory Comput. 14, 6269–6275 (2018). DOI: 10.1021/acs.jctc.8b00728
We report first principles calculations of electronic gaps, lifetimes and photoelectron spectra of a series of molecules, performed by efficiently combining the computation of electron-electron and electron-phonon self-energies. The dielectric matrix is represented in terms of dielectric eigenpotentials, utilized for both the calculation of $G_0W_0$ quasi-particle energies and the diagonalization of the dynamical matrix; virtual electronic states are never explicitly computed and all self-energies are evaluated over the full frequency spectrum. Our formulation enables electronic structure calculations at the many-body perturbation theory level, inclusive of electron-phonon coupling, for systems with hundreds of electrons.
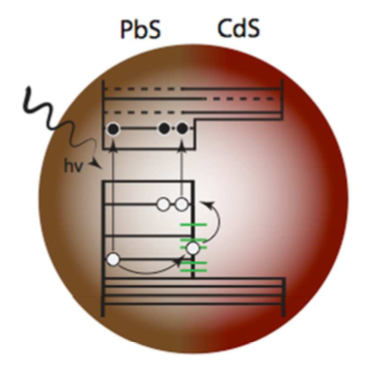
Enhanced Multiple Exciton Generation in PbS|CdS Janus-Like Heterostructured Nanocrystals
Daniel M. Kroupa, Gregory F. Pach, Márton Vörös, Federico Giberti, Boris D Chernomordik, Ryan W. Crisp, Arthur J Nozik, Justin C. Johnson, Rohan Singh, Victor I. Klimov, Giulia Galli, and Matthew C. Beard
ACS Nano 12, 10084–10094 (2018). DOI: 10.1021/acsnano.8b04850
Generating multiple excitons by a single high-energy photon is a promising third generation solar energy conversion strategy. We demonstrate that multiple exciton generation (MEG) in PbS|CdS Janus-like hetero-nanostructures is enhanced over that of single-component and core/shell nanocrystal architectures, with an onset close to two times the PbS band gap. We attribute the enhanced MEG to the asymmetric nature of the hetero-nanostructure that results in an increase in the effective Coulomb interaction that drives MEG and a reduction of the competing hot exciton cooling rate. Slowed cooling occurs through effective trapping of hot- holes by a manifold of valence band interfacial states having both PbS and CdS character, as evidenced by photoluminescence studies and by ab initio calculations. Using transient photocurrent spectroscopy, we find that the MEG characteristics of the individual nanostructures are maintained in conductive arrays and demonstrate that these quasi-spherical PbS|CdS nanocrystals can be incorporated as the main absorber layer in functional solid-state solar cell architectures. Finally, based upon our analysis, we provide design rules for the next generation of engineered nanocrystals to further improve the MEG characteristics.
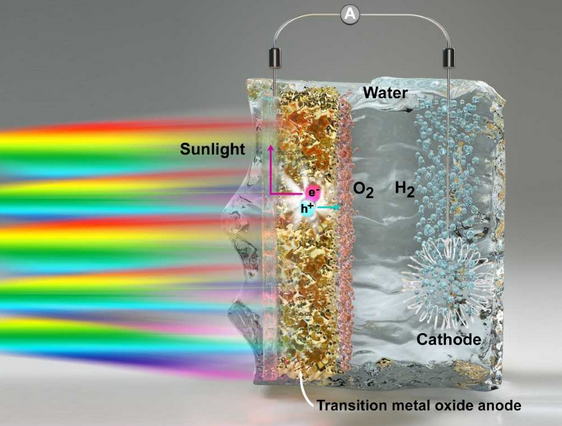
The role of defects and excess surface charges at finite temperature for optimizing oxide photoabsorbers
Matteo Gerosa, Francois Gygi, Marco Govoni, and Giulia Galli
Nat. Mater. 17, 1122-1127 (2018). DOI: 10.1038/s41563-018-0192-4
Computational screening of materials for solar to fuel conversion technologies have mostly focused on bulk properties, thus neglecting the structure and chemistry of surfaces and interfaces with water. We report a finite temperature study of WO3, a promising anode for photo-electrochemical cells, carried out using first principles molecular dynamics simulations coupled with many body perturbation theory. We identified three major factors determining the chemical reactivity of the material in- terfaced with water: the presence of surface defects, the dynamics of excess charge at the surface, and finite temperature fluctuations of the surface electronic orbitals. These general descriptors are essential for the understanding and prediction of opti- mal oxide photo-absorbers for water oxidation.
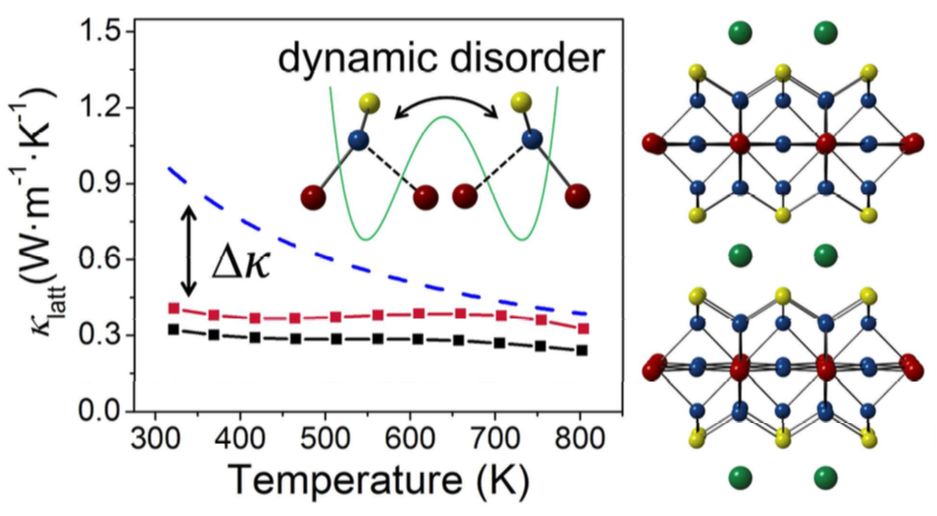
Two-Dimensional CsAg5Te3-xSx Semiconductors: Multi-Anion Chalcogenides with Dynamic Disorder and Ultralow Thermal Conductivity
James M Hodges, Yi Xia, Christos D. Malliakas, Grant C. B. Alexander, Maria K. Y. Chan, and Mercouri G. Kanatzidis
Chem. Mater. 30, 7245-7254 (2018). DOI: 10.1021/acs.chemmater.8b03306
Metal chalcogenides underpin a wide variety of energy-related applications and are ideal systems for probing lattice dynamics and fundamental transport phenomena. Here, we describe the synthesis and transport properties of CsAg5TeS2 and its solid solution CsAg5Te3-xSx (x = 1-2), new semiconductors with tunable bandgaps ranging from 0.17 to 0.30 eV. CsAg5TeS2 has a fully ordered two-dimensional structure that includes a group of Ag atoms in a heteroleptic, tetrahedral coordination geometry (AgTe2S2). Single crystal X-ray diffraction indicates that the compounds crystallize in the tetragonal space group P4/mmm, while pair-distribution function (PDF) analysis reveals off-centering at the heteroleptic Ag sites, signifying a lower symmetry I4/mcm space group. The underlying disorder acts as a phonon-blocking mechanism that helps facilitate ultralow lattice thermal conductivity below 0.40 W⋅m-1⋅K-1 at ~300 K and highlights the importance of local disorder in thermal transport. Density functional theory (DFT) provides additional insight into the electronic and thermal properties of the materials, which are good candidates for p-type thermoelectrics.
signac: A Python framework for data and workflow management
Vyas Ramasubramani, Carl S. Adorf, Paul M. Dodd, Bradley D. Dice, Sharon C. Glotzer
Proc. 17th Python Sci. Conf. 152–159 (2018). DOI: 10.25080/Majora-4af1f417-016
Computational research requires versatile data and workflow man- agement tools that can easily adapt to the highly dynamic requirements of scientific investigations. Many existing tools require strict adherence to a par- ticular usage pattern, so researchers often use less robust ad hoc solutions that they find easier to adopt. The resulting data fragmentation and methodological incompatibilities significantly impede research. Our talk showcases signac, an open-source Python framework that offers highly modular and scalable solutions for this problem. Named for the Pointillist painter Paul Signac, the framework’s powerful workflow management tools enable users to construct and automate workflows that transition seamlessly from laptops to HPC clusters. Crucially, the underlying data model is completely independent of the workflow. The flexible, serverless, and schema-free signac database can be introduced into other workflows with essentially no overhead and no recourse to the signac workflow model. Additionally, the data model’s simplicity makes it easy to parse the underlying data without using signac at all. This modularity and simplicity eliminates significant barriers for consistent data management across projects, facilitating improved provenance management and data sharing with minimal overhead.
Revisiting lattice thermal transport in PbTe: The crucial role of quartic anharmonicity
Yi Xia
App. Phys. Lett. 113, 073901 (2018). DOI: 10.1063/1.5040887
We perform a first-principles study of lattice thermal transport in PbTe by explicitly considering anharmonicity up to 4th order. To determine the temperature-dependent lattice constant of PbTe beyond quasiharmonic approximation, we introduce a simple yet effective scheme to account for anharmonic phonon renormalization at finite temperature. Moreover, we explicitly compute mode-resolved phonon lifetimes by including both three- and four-phonon scatterings. We find that (1) anharmonic phonon renormalization leads to strong vibrational frequency shifts which improve the agreement between simulated and experimental lattice constants; (2) these frequency shifts lead to a significant increase in lattice thermal conductivity (κl) because of reduced phonon scattering phase space; and (3) four-phonon scatterings are responsible for severe reduction in κl on top of three-phonon scatterings, making κl consistent with experiments. Our results suggest that the predicted κl and its temperature dependence without considering thermal expansion, anharmonic phonon renormalization and four-phonon scatterings could accidentally agree with experiments due to error cancellation. Our study not only deepens the understanding of lattice thermal transport in PbTe but also exemplifies a widely applicable approach to investigate lattice dynamics and thermal transport properties from first-principles calculations including high-order anharmonicity.
On the Relation between Equation-of-Motion Coupled-Cluster Theory and the GW Approximation
Malte Lange and Timothy Berkelbach
J. Chem. Theory Comput. 14, 4224-4236 (2018). DOI: 10.1021/acs.jctc.8b00455
We discuss the analytic and diagrammatic structure of ionization potential (IP) and electron affinity (EA) equation-of-motion coupled-cluster (EOM-CC) theory, in order to put it on equal footing with the prevalent GW approximation. The comparison is most straightforward for the time-ordered one-particle Green’s function, and we show that the Green’s function calculated by EOM-CC with single and double excitations (EOM-CCSD) includes fewer ring diagrams at higher order than does the GW approximation, due to the former’s unbalanced treatment of time-ordering. However, the EOM-CCSD Green’s function contains a large number of vertex corrections, including ladder diagrams, mixed ring-ladder diagrams, and exchange diagrams. By including triple excitations, the EOM-CCSDT Green’s function includes all diagrams contained in the GW approximation, along with many high-order vertex corrections. In the same language, we discuss a number of common approximations to the EOM-CCSD equations, many of which can be classified as elimination of diagrams. Finally, we present numerical results by calculating the principal charged excitations energies of the molecules contained in the so-called GW100 test set [ J. Chem. Theory Comput. 2015, 11, 5665−5687]. We argue that (in molecules) exchange is as important as screening, advocating for a Hartree–Fock reference and second-order exchange in the self-energy.
Dielectric properties of condensed systems composed of fragments
Ding Pan, Marco Govoni, and Giulia Galli
J. Chem. Phys. 149, 051101 (2018). DOI: 10.1063/1.5044636
The dielectric properties of molecules and nanostructures are usually modified in a complex manner, when assembled into a condensed phase. We propose a first-principles method to compute polarizabilities of sub-entities of solids and liquids, which accounts for multipolar interactions at all orders and is applicable to semiconductors and insulators. The method only requires the evaluation of induced fields in the condensed phase, with no need of multiple calculations for each constituent. As an example, we present results for the molecular polarizabilities of water in a wide pressure and temperature range. We found that at ambient conditions, the dipole-induced-dipole approximation is sufficiently accurate and the Clausius-Mossotti relation may be used, e.g., to obtain molecular polarizabilities from experimental refractive indexes. However with increasing pressure, this approximation becomes unreliable and in the case of ice X the Clausius-Mossotti relation is not valid.
Layered nested Markov chain Monte Carlo
Nicholas E. Jackson, Michael A. Webb, and Juan J. de Pablo
J. Chem. Phys. 149, 072326 (2018). DOI: 10.1063/1.5030531
A configurational sampling algorithm based on nested layerings of Markov chains (Layered Nested Markov Chain Monte Carlo or L-NMCMC) is presented for simulations of systems characterized by rugged free energy landscapes. The layerings are generated using a set of auxiliary potential energy surfaces. The implementation of the method is demonstrated in the context of a rugged, two-dimensional potential energy surface. The versatility of the algorithm is next demonstrated on a simple, many-body system, namely, a canonical Lennard-Jones fluid in the liquid state. In that example, different layering schemes and auxiliary potentials are used, including variable cutoff distances and excluded-volume tempering. In addition to calculating a variety of properties of the system, it is also shown that L-NMCMC, when combined with a free-energy perturbation formalism, provides a straightforward means to construct approximate free-energy surfaces at no additional computational cost using the sampling distributions of each auxiliary Markov chain. The proposed L-NMCMC scheme is general in that it could be complementary to any number of methods that rely on sampling from a target distribution or methods that exploit a hierarchy of time scales and/or length scales through decomposition of the potential energy.

Evolutionary strategy for inverse charge measurements of dielectric particles
Xikai Jiang, Jiyuan Li, Victor Lee, Heinrich M. Jaeger, Olle G. Heinonen, and Juan J. de Pablo
J. Chem. Phys. 148, 234302 (2018). DOI: 10.1063/1.5027435
We report a computational strategy to obtain the charges of individual dielectric particles from experimental observation of their interactions as a function of time. This strategy uses evolutionary optimization to minimize the difference between trajectories extracted from experiment and simulated trajectories based on many-particle force fields. The force fields include both Coulombic interactions and dielectric polarization effects that arise due to particle-particle charge mismatch and particle-environment dielectric contrast. The strategy was applied to systems of free falling charged granular particles in vacuum, where electrostatic interactions are the only driving forces that influence the particles' motion. We show that when the particles' initial positions and velocities are known, the optimizer requires only an initial and final particle configuration of a short trajectory in order to accurately infer the particles' charges; when the initial velocities are unknown and only the initial positions are given, the optimizer can learn from multiple frames along the trajectory to determine the particles' initial velocities and charges. While the results presented here offer a proof-of-concept demonstration of the proposed ideas, the proposed strategy could be extended to more complex systems of electrostatically charged granular matter.
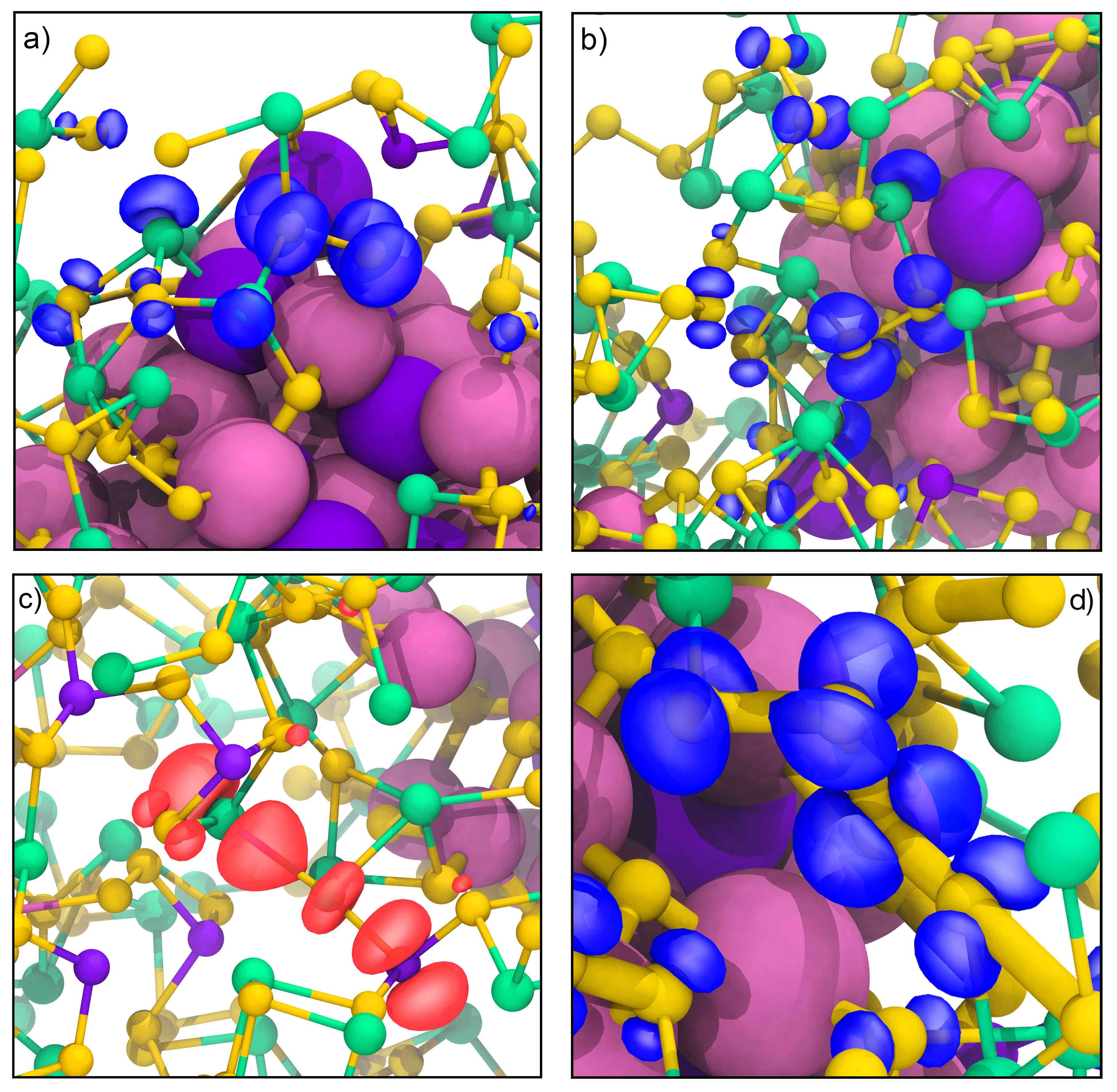
Surface chemistry and buried interfaces in all-inorganic nanocrystalline solids
Emilio Scalise, Vishwas Srivastava, Eric M. Janke, Dmitri Talapin, Giulia Galli, and Stefan Wipperman,
Nat. Nanotech. 13, 841-848 (2018). DOI: 10.1038/s41565-018-0189-9
Semiconducting nanomaterials synthesized using wet chemical techniques play an important role in emerging optoelectronic and photonic technologies. Controlling the surface chemistry of the nano building blocks and their interfaces with ligands is one of the outstanding challenges for the rational design of these systems. We present an integrated theoretical and experimental approach to characterize, at the atomistic level, buried interfaces in solids of InAs nanoparticles capped with Sn2S64– ligands. These prototypical nanocomposites are known for their promising transport properties and unusual negative photoconductivity. We found that inorganic ligands dissociate on InAs to form a surface passivation layer. A nanocomposite with unique electronic and transport properties is formed, that exhibits type II heterojunctions favourable for exciton dissociation. We identified how the matrix density, sulfur content and specific defects may be designed to attain desirable electronic and transport properties, and we explain the origin of the measured negative photoconductivity of the nanocrystalline solids.

Ab Initio Spectroscopy and Ionic Conductivity of Water under Earth Mantle Conditions
Viktor Rozsa, Ding Pan, Federico Giberti, and Giulia Galli
PNAS 115, 6952-6957 (2018). DOI: 10.1073/pnas.1800123115
The phase diagram of water at extreme conditions plays a critical role in Earth and planetary science, yet remains poorly understood. Here we report a first-principles investigation of the liquid at high temperature, between 11 GPa and 20 GPa—a region where numerous controversial results have been reported over the past three decades. Our results are consistent with the recent estimates of the water melting line below 1,000 K and show that on the 1,000-K isotherm the liquid is rapidly dissociating and recombining through a bimolecular mechanism. We found that short-lived ionic species act as charge carriers, giving rise to an ionic conductivity that at 11 GPa and 20 GPa is six and seven orders of magnitude larger, respectively, than at ambient conditions. Conductivity calculations were performed entirely from first principles, with no a priori assumptions on the nature of charge carriers. Despite frequent dissociative events, we observed that hydrogen bonding persists at high pressure, up to at least 20 GPa. Our computed Raman spectra, which are in excellent agreement with experiment, show no distinctive signatures of the hydronium and hydroxide ions present in our simulations. Instead, we found that infrared spectra are sensitive probes of molecular dissociation, exhibiting a broad band below the OH stretching mode ascribable to vibrations of complex ions.
Efficient encapsulation of proteins with random copolymers
Trung Dac Nguyen, Baofu Qiao, and Monica Olvera de la Cruz
PNAS 115, 6578-6583 (2018). DOI: 10.1073/pnas.1806207115
Membraneless organelles are aggregates of disordered proteins that form spontaneously to promote specific cellular functions in vivo. The possibility of synthesizing membraneless organelles out of cells will therefore enable fabrication of protein-based materials with functions inherent to biological matter. Since random copolymers contain various compositions and sequences of solvophobic and solvophilic groups, they are expected to function in nonbiological media similarly to a set of disordered proteins in membraneless organelles. Interestingly, the internal environment of these organelles has been noted to behave more like an organic solvent than like water. Therefore, an adsorbed layer of random copolymers that mimics the function of disordered proteins could, in principle, protect and enhance the proteins’ enzymatic activity even in organic solvents, which are ideal when the products and/or the reactants have limited solubility in aqueous media. Here, we demonstrate via multiscale simulations that random copolymers efficiently incorporate proteins into different solvents with the potential to optimize their enzymatic activity. We investigate the key factors that govern the ability of random copolymers to encapsulate proteins, including the adsorption energy, copolymer average composition, and solvent selectivity. The adsorbed polymer chains have remarkably similar sequences, indicating that the proteins are able to select certain sequences that best reduce their exposure to the solvent. We also find that the protein surface coverage decreases when the fluctuation in the average distance between the protein adsorption sites increases. The results herein set the stage for computational design of random copolymers for stabilizing and delivering proteins across multiple media.
First-Principles Simulations of Liquid Water Using a Dielectric-Dependent Hybrid Functional
Alex Gaiduk, Jeffrey Gustafson, Francois Gygi, and Giulia Galli
J. Phys. Chem. Lett. 9, 3068-3073 (2018). DOI: 10.1021/acs.jpclett.8b01017
We carried out first-principles simulations of liquid water under ambient conditions using a dielectric-dependent hybrid functional, where the fraction of exact exchange is set equal to the inverse of the high-frequency dielectric constant of the liquid. We found excellent agreement with experiment for the oxygen–oxygen partial correlation function at the experimental equilibrium density and 311 ± 3 K. Other structural and dynamical properties, such as the diffusion coefficient, molecular dipole moments, and vibrational spectra, are also in good agreement with experiment. Our results, together with previous findings on electronic properties of the liquid with the same functional, show that the dielectric-dependent hybrid functional accurately describes both the structural and electronic properties of liquid water.
Lattice thermal transport in group II-alloyed PbTe
Yi Xia, James M. Hodges, Mercouri G. Kanatzidis, and Maria K. Y. Chan
Appl. Phys. Lett. 112, 181906 (2018). DOI: 10.1063/1.5002587
PbTe, one of the most promising thermoelectric materials, has recently demonstrated a thermoelectric figure of merit (ZT) of above 2.0 when alloyed with group II elements. The improvements are due mainly to significant reduction of lattice thermal conductivity (κl), which was in turn attributed to nanoparticle precipitates. However, a fundamental understanding of various phonon scattering mechanisms within the bulk alloy is still lacking. In this work, we apply the newly-developed density-functional-theory-based compressive sensing lattice dynamics approach to model lattice heat transport in PbTe, MTe, and Pb0.94M0.06Te (M = Mg, Ca, Sr, and Ba) and compare our results with experimental measurements, with focus on the strain effect and mass disorder scattering. We find that (1) CaTe, SrTe, and BaTe in the rock-salt structure exhibit much higher κl than PbTe, while MgTe in the same structure shows anomalously low κl; (2) lattice heat transport of PbTe is extremely sensitive to static strain induced by alloying atoms in solid solution form; (3) mass disorder scattering plays a major role in reducing κl for Mg/Ca/Sr-alloyed PbTe through strongly suppressing the lifetimes of intermediate- and high-frequency phonons, while for Ba-alloyed PbTe, precipitated nanoparticles are also important.
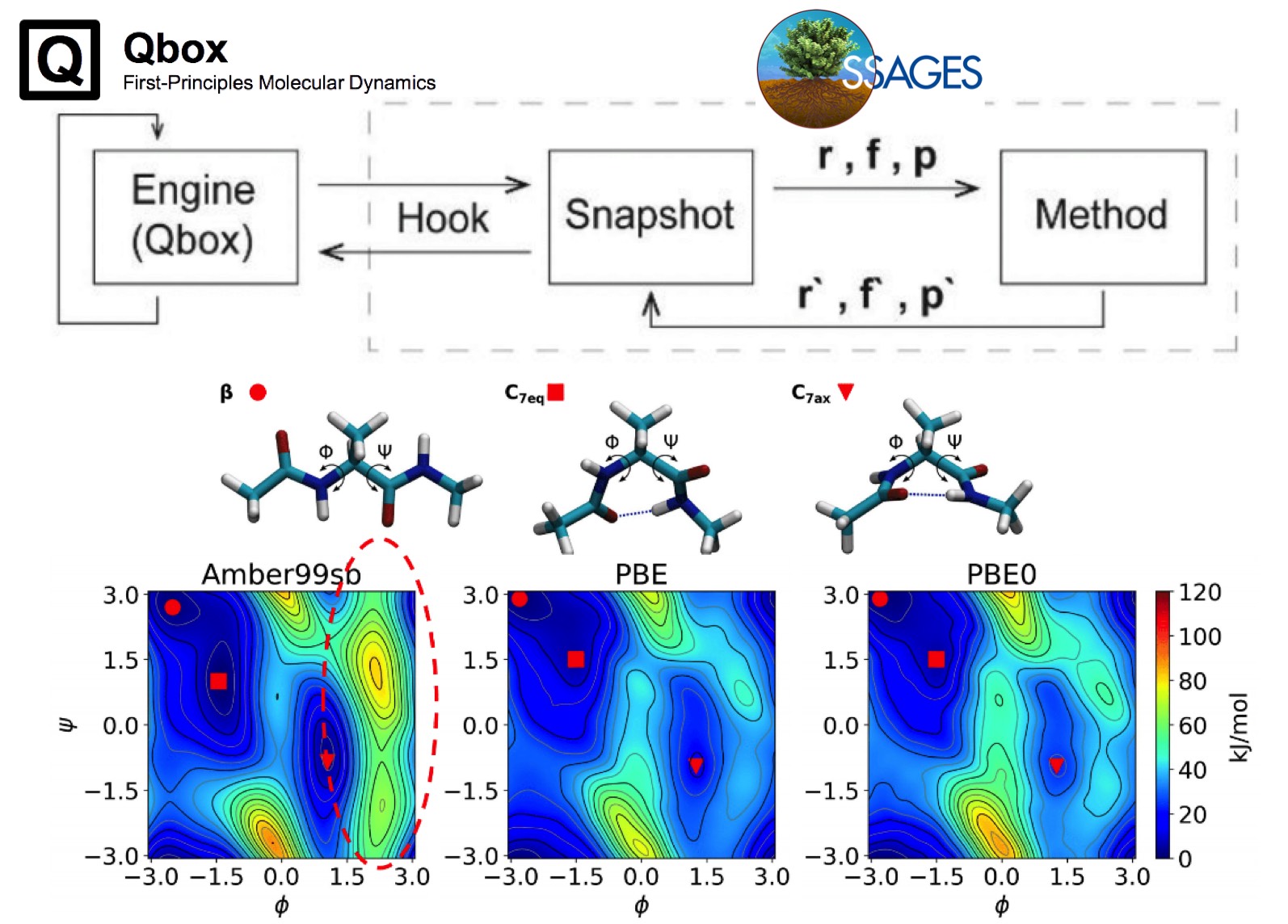
Hierarchical Coupling of First Principles Molecular Dynamics with Advanced Sampling Methods
Emre Sevgen, Federico Giberti, Hythem Sidky, Jonathan Whitmer, Giulia Galli, Francois Gygi, and Juan de Pablo
J. Chem. Theory Comput. 14, 2881–2888 (2018). DOI: 10.1021/acs.jctc.8b00192
We present a seamless coupling of a suite of codes designed to perform advanced sampling simulations, with a first-principles molecular dynamics (MD) engine. As an illustrative example, we discuss results for the free energy and potential surfaces of the alanine dipeptide obtained using both local and hybrid density functionals (DFT), and we compare them with those of a widely used classical force field, Amber99sb. In our calculations, the efficiency of first-principles MD using hybrid functionals is augmented by hierarchical sampling, where hybrid free energy calculations are initiated using estimates obtained with local functionals. We find that the free energy surfaces obtained from classical and first-principles calculations differ. Compared to DFT results, the classical force field overestimates the internal energy contribution of high free energy states, and it underestimates the entropic contribution along the entire free energy profile. Using the string method, we illustrate how these differences lead to different transition pathways connecting the metastable minima of the alanine dipeptide. In larger peptides, those differences would lead to qualitatively different results for the equilibrium structure and conformation of these molecules.
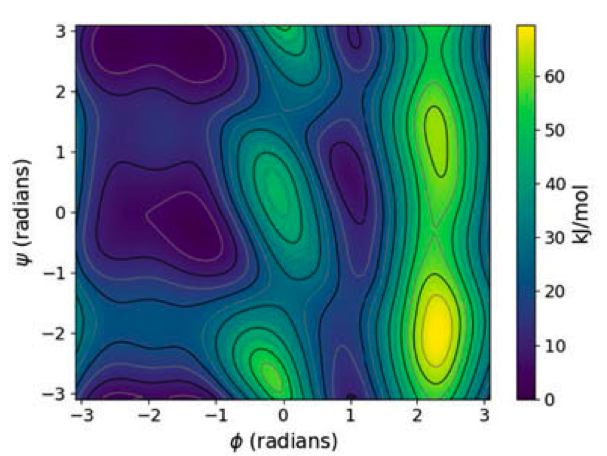
Adaptive enhanced sampling by force-biasing using neural networks
Ashley Z. Guo, Emre Sevgen, Hythem Sidky, Jonathan K. Whitmer, Jeffrey A. Hubbell, and Juan J. de Pablo
J. Chem. Phys. 148, 134108 (2018). DOI: 10.1063/1.5020733
A machine learning assisted method is presented for molecular simulation of systems with rugged free energy landscapes. The method is general and can be combined with other advanced sampling techniques. In the particular implementation proposed here, it is illustrated in the context of an adaptive biasing force approach where, rather than relying on discrete force estimates, one can resort to a self-regularizing artificial neural network to generate continuous, estimated generalized forces. By doing so, the proposed approach addresses several shortcomings common to adaptive biasing force and other algorithms. Specifically, the neural network enables (1) smooth estimates of generalized forces in sparsely sampled regions, (2) force estimates in previously unexplored regions, and (3) continuous force estimates with which to bias the simulation, as opposed to biases generated at specific points of a discrete grid. The usefulness of the method is illustrated with three different examples, chosen to highlight the wide range of applicability of the underlying concepts. In all three cases, the new method is found to enhance considerably the underlying traditional adaptive biasing force approach. The method is also found to provide improvements over previous implementations of neural network assisted algorithms.
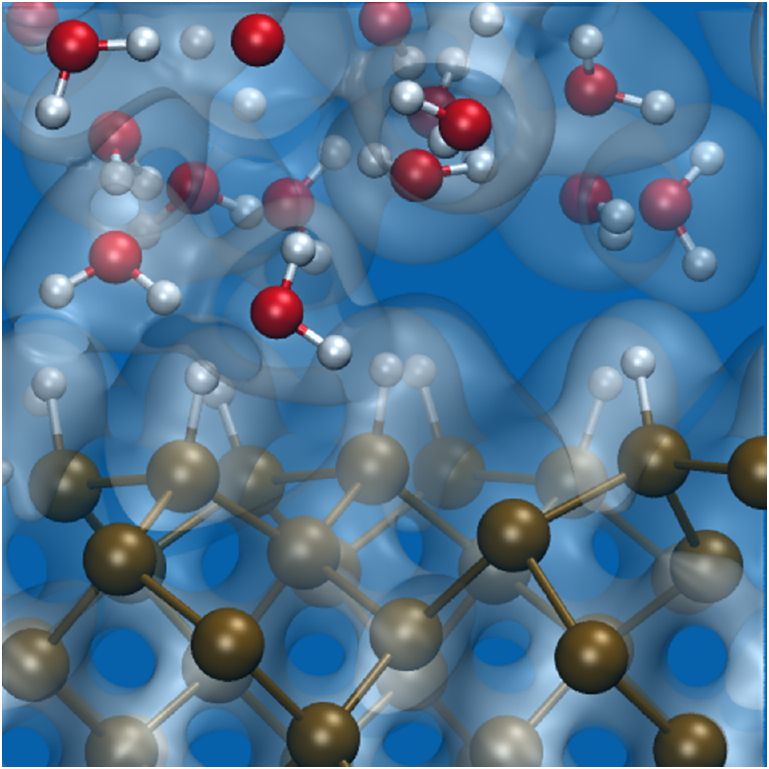
Roadmap on semiconductor–cell biointerfaces
Bozhi Tian, Shuai Xu, John A Rogers, Stefano Cestellos-Blanco, Peidong Yang, João L Carvalho-de-Souza, Francisco Bezanilla, Jia Liu, Zhenan Bao, Martin Hjort, Yuhong Cao, Nicholas Melosh, Guglielmo Lanzani, Fabio Benfenati, Giulia Galli, Francois Gygi, Rylan Kautz, Alon A Gorodetsky, Samuel S Kim, Timothy K Lu, Polina Anikeeva, Michal Cifra, Ondrej Krivosudský, Daniel Havelka, and Yuanwen Jiang
Phys. Biol. 15, 031002 (2018). DOI: 10.1088/1478-3975/aa9f34
This roadmap outlines the role semiconductor-based materials play in understanding the complex biophysical dynamics at multiple length scales, as well as the design and implementation of next-generation electronic, optoelectronic, and mechanical devices for biointerfaces. The roadmap emphasizes the advantages of semiconductor building blocks in interfacing, monitoring, and manipulating the activity of biological components, and discusses the possibility of using active semiconductor–cell interfaces for discovering new signaling processes in the biological world.
Equilibration and analysis of first-principles molecular dynamics simulations of water
William Dawson, Francois Gygi
J. Chem. Phys. 148, 124501 (2018). DOI: 10.1063/1.5018116
First-principles molecular dynamics (FPMD) simulations based on density functional theory are becoming increasingly popular for the description of liquids. In view of the high computational cost of these simulations, the choice of an appropriate equilibration protocol is critical. We assess two methods of estimation of equilibration times using a large dataset of first-principles molecular dynamics simulations of water. The Gelman-Rubin potential scale reduction factor [A. Gelman and D. B. Rubin, Stat. Sci. 7, 457 (1992)] and the marginal standard error rule heuristic proposed by White [Simulation 69, 323 (1997)] are evaluated on a set of 32 independent 64-molecule simulations of 58 ps each, amounting to a combined cumulative time of 1.85 ns. The availability of multiple independent simulations also allows for an estimation of the variance of averaged quantities, both within MD runs and between runs. We analyze atomic trajectories, focusing on correlations of the Kohn-Sham energy, pair correlation functions, number of hydrogen bonds, and diffusion coefficient. The observed variability across samples provides a measure of the uncertainty associated with these quantities, thus facilitating meaningful comparisons of different approximations used in the simulations. We find that the computed diffusion coefficient and average number of hydrogen bonds are affected by a significant uncertainty in spite of the large size of the dataset used. A comparison with classical simulations using the TIP4P/2005 model confirms that the variability of the diffusivity is also observed after long equilibration times. Complete atomic trajectories and simulation output files are available online for further analysis.
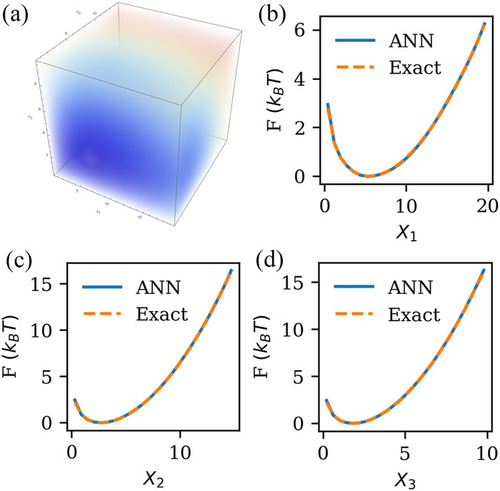
Learning free energy landscapes using artificial neural networks
Hythem Sidky, Jonathan Whitmer
J. Chem. Phys. 148, 104111 (2018). DOI: 10.1063/1.5018708
Existing adaptive bias techniques, which seek to estimate free energies and physical properties from molecular simulations, are limited by their reliance on fixed kernels or basis sets which hinder their ability to efficiently conform to varied free energy landscapes. Further, user-specified parameters are in general non-intuitive yet significantly affect the convergence rate and accuracy of the free energy estimate. Here we propose a novel method, wherein artificial neural networks (ANNs) are used to develop an adaptive biasing potential which learns free energy landscapes. We demonstrate that this method is capable of rapidly adapting to complex free energy landscapes and is not prone to boundary or oscillation problems. The method is made robust to hyperparameters and overfitting through Bayesian regularization which penalizes network weights and auto-regulates the number of effective parameters in the network. ANN sampling represents a promising innovative approach which can resolve complex free energy landscapes in less time than conventional approaches while requiring minimal user input.
Simple data and workflow management with the Signac framework
Carl S. Adorf, Paul M. Dodd, Vyas Ramasubramani, Sharon C. Glotzer
Comp. Mater. Sci. 146, 220-229 (2018). DOI: 10.1016/j.commatsci.2018.01.035com
Researchers in the fields of materials science, chemistry, and computational physics are regularly posed with the challenge of managing large and heterogeneous data spaces. The amount of data increases in lockstep with computational efficiency multiplied by the amount of available computational resources, which shifts the bottleneck in the scientific process from data acquisition to data processing and analysis. We present a framework designed to aid in the integration of various specialized data formats, tools and workflows. The signac framework provides all basic components required to create a well-defined and thus collectively accessible and searchable data space, simplifying data access and modification through a homogeneous data interface that is largely agnostic to the data source, i.e., computation or experiment. The framework’s data model is designed to not require absolute commitment to the presented implementation, simplifying adaption into existing data sets and workflows. This approach not only increases the efficiency with which scientific results can be produced, but also significantly lowers barriers for collaborations requiring shared data access.

Weak Polyelectrolyte Complexation Driven by Associative Charging
Vikramjit S. Rathee, Aristotle J. Zervoudakis, Hythem Sidky, Benjamin J. Sikora and Jonathan K. Whitmer
J. Chem. Phys. 148, 114901 (2018). DOI: 10.1063/1.5017941
Weak polyelectrolytes are relevant for a wide range of fields; in particular, they are useful in the creation of “smart” materials for chemical separations and drug delivery. The charges on weak polyelectrolytes are dynamic and sensitive to the surroundings, causing polymer chains to adopt environmentally-responsive conformations. Currently, there exists no comprehensive picture of this behavior, particularly where polymer–polymer interactions have the potential to affect charging properties significantly. In this study, we elucidate the novel interplay between weak polyelectrolyte charging and complexation behavior through coupled molecular dynamics and Monte Carlo simulations. Specifically, we investigate a model of two equal-length and oppositely charging polymer chains in implicit salt solution represented through Debye–Hückel interactions. The charging tendency of each chain, along with the salt concentration, are varied to determine the existence and extent of cooperativity in charging and complexation. Strong cooperation in the charging of these chains is observed at large Debye lengths, corresponding to low salt concentrations, while at lower Debye lengths (higher salt concentrations), the chains behave in apparent isolation. When the electrostatic coupling is long-ranged, we find that a highly charged chain strongly promotes the charging of its partner chain, even if the environment is unfavorable for an isolated version of that partner chain. Evidence of this phenomenon is supported by a drop in the potential energy of the system, which does not occur at the lower Debye lengths where both potential energies and charge fractions converge for all partner chain charging tendencies. The discovery of this cooperation will be helpful in developing “smart” drug delivery mechanisms by allowing for better predictions for the dissociation point of delivery complexes.
In silico measurement of elastic moduli of nematic liquid crystals
Hythem Sidky, Juan J. de Pablo, and Jonathan K. Whitmer
Phys. Rev. Lett. 120, 107801 (2018). DOI: 10.1103/PhysRevLett.120.107801
Experiments on confined droplets of the nematic liquid crystal 5CB have questioned long- established bounds imposed on the elastic free energy of nematic systems. This elasticity, which derives from molecular alignment within nematic systems, is quantified through a set of moduli which can be difficult to measure experimentally and, in some cases, can only be probed indirectly. This is particularly true of the surface-like saddle-splay elastic term, for which the available ex- perimental data indicate values on the cusp of stability, often with large uncertainties. Here, we demonstrate that all nematic elastic moduli, including the saddle-splay elastic constant k24, may be calculated directly from atomistic molecular simulation. Importantly, results obtained through in silico measurements of the 5CB elastic properties demonstrate unambiguously that saddle-splay elasticity alone is unable to describe the observed confined morphologies.
Ion Distribution in Microphase-Separated Copolymers with Periodic Dielectric Permittivity
Weiwei Chu, Jian Qin, and Juan J. de Pablo
Macromolecules 51, 1986–1991 (2018). DOI: 10.1021/acs.macromol.7b02508
We examine the distribution of ionic species in charged, microphase-separated diblock copolymers by relying on coarse-grained simulations of the underlying materials. The model adopted here has been particularly useful in understanding the behavior of neutral block polymer systems. In this work, it is extended to describe charged molecules. A simulation methodology is proposed in which dielectric inhomogeneities within the phase-separated systems are taken into account by solving on-the-fly Poisson’s equation for spatially varying dielectric permittivity and calculating the corresponding electric fields. The Green’s function appropriate for a periodic and ionic system is explicitly calculated and used in simulations to arrive at phase diagrams as a function of salt concentration and copolymer composition. The systems considered here are representative of lithium salt-doped diblock copolymers, which could be of potential use for solid-electrolyte batteries.

GW100: Comparison of Methods and Accuracy of Results Obtained with the WEST Code
Marco Govoni and Giulia Galli
J. Chem. Theory Comput. 14, 1895–1909 (2018). DOI: 10.1021/acs.jctc.7b00952
The reproducibility of calculations carried out within many-body perturbation the- ory at the G0W0 level is assessed for 100 closed shell molecules and compared to that of density functional theory. We consider vertical ionization potentials (VIP) and electron affinities (VEA) obtained with five different codes: BerkeleyGW, FHI-aims, TURBOMOLE, VASP, and WEST. We review the approximations and parameters that control the accuracy of G0W0 results in each code and we discuss in detail the effect of extrapolation techniques for the parameters entering the WEST code. Differences between the VIP and VEA computed with the various codes are within ∼ 60 meV and ∼ 120 meV respectively, which is up to four times larger than in the case of the best results obtained with DFT codes. Vertical ionization potentials are validated against experiment and CCSD(T) quantum chemistry results showing a mean absolute relative error of ∼ 4% for data obtained with WEST. Our analysis of the differences between localized orbitals and plane-wave implementations points out at molecules containing Cu, I, Ga, and Xe as major sources of discrepancies, which call for a re-evaluation of the pseudopotentials used for these systems in G0W0 calculations.

Electron affinity of liquid water
Alex Gaiduk, T. Anh Pham, Marco Govoni, Francesco Paesani and Giulia Galli
Natur. Commun. 9, 237 (2018). DOI: 10.1038/s41467-017-02673-z
Understanding redox and photochemical reactions in aqueous environments requires a precise knowledge of the ionization potential and electron affinity of liquid water. The former has been measured, but not the latter. We predict the electron affinity of liquid water and of its surface from first principles, coupling path-integral molecular dynamics with ab initio potentials, and many-body perturbation theory. Our results for the surface (0.8 eV) agree well with recent pump-probe spectroscopy measurements on amorphous ice. Those for the bulk (0.1–0.3 eV) differ from several estimates adopted in the literature, which we critically revisit. We show that the ionization potential of the bulk and surface are almost identical; instead their electron affinities differ substantially, with the conduction band edge of the surface much deeper in energy than that of the bulk. We also discuss the significant impact of nuclear quantum effects on the fundamental gap and band edges of the liquid.

Charge Transport and Phase Behavior of Imidazolium-Based Ionic Liquid Crystals from Fully Atomistic Simulations
Michael Quevillon and Jonathan K. Whitmer
Materials 11, 64 (2018). DOI: 10.3390/ma11010064
Ionic liquid crystals occupy an intriguing middle ground between room-temperature ionic liquids and mesostructured liquid crystals. Here, we examine a non-polarizable, fully atomistic model of the 1-alkyl-3-methylimidazolium nitrate family using molecular dynamics in the constant pressure–constant temperature ensemble. These materials exhibit a distinct “smectic” liquid phase, characterized by layers formed by the molecules, which separate the ionic and aliphatic moieties. In particular, we discuss the implications this layering may have for electrolyte applications.
Simulating the Thermodynamics of Charging in Weak Polyelectrolytes: The Debye Hückel Limit
Vikramjit S. Rathee, Benjamin J. Sikora, Hythem Sidky and Jonathan K. Whitmer
Materials Research Express 5, 014010 (2018). DOI: 10.1088/2053-1591/aaa049
The coil–globule transition in weak (annealed) polyelectrolytes involves a subtle balance of pH, charge strength, and solvation forces. In this work, we utilize a coarse-grained hybrid grand-canonical Monte Carlo and molecular dynamics approach to explore the swelling behavior of weak linear and star polyelectrolytes under different ionic screening conditions and pH. Importantly, we are able to quantify topology-dependent effects in charging which arise at the core of star polymers. Our results are suggestive of suppression of charging in star weak polyelectrolytes in comparison to linear weak polyelectrolytes. Furthermore, we characterize the coil–globule transition in linear and star weak polyelectrolyte through expanded ensemble density-of-states simulations which suggest a change from a first order to second order phase transition moving from linear to star polyelectrolytes. Lastly, we characterize the inhomogeneous charging across the weak star polyelectrolyte through observed shifts in ${\rm{\Delta }}{{\rm{pK}}}_{{\rm{o}}},$ and compare with experimental work. We discuss these results in relation to surfaces functionalized by weak polyelectrolyte brushes and weak polyelectrolyte-based drug delivery applications
Low-temperature anomalies of a vapor deposited glass
Beatriz Seoane, Daniel R. Reid, Juan J. de Pablo, and Francesco Zamponi
Phys. Rev. Materials 2, 015602 (2018). DOI: 10.1103/PhysRevMaterials.2.015602
We investigate the low-temperature properties of two-dimensional Lennard-Jones glass films, prepared in silico both by liquid cooling and by physical vapor deposition. We identify deep in the solid phase a crossover temperature T∗, at which slow dynamics and enhanced heterogeneity emerge. Around T∗, localized defects become visible, leading to vibrational anomalies as compared to standard solids. We find that, on average, T∗ decreases in samples with lower inherent structure energy, suggesting that such anomalies will be suppressed in ultrastable glass films, prepared both by very slow liquid cooling and vapor deposition.
SSAGES: Software Suite for Advanced General Ensemble Simulations
Hythem Sidky, Yamil J. Colon, Julian Helfferich, Benjamin J. Sikora, Cody Bezik, Weiwei Chu, Federico Giberti, Ashley Z. Guo, Xikai Jiang, Joshua Lequieu, Jiyuan Li, Joshua Moller, Michael J. Quevillon, Mohammad Rahimi, Hadi Ramezani-Dakhel, Vikramjit S. Rathee, Daniel R. Reid, Emre Sevgen, Vikram Thapar, Michael A. Webb, Jonathan K. Whitmer, and Juan J. de Pablo
J. Chem. Phys. 148, 044104 (2018). DOI: 10.1063/1.5008853
Molecular simulation has emerged as an essential tool for modern-day research, but obtaining proper results and making reliable conclusions from simulations requires adequate sampling of the system under consideration. To this end, a variety of methods exist in the literature that can enhance sampling considerably, and increasingly sophisticated, effective algorithms continue to be developed at a rapid pace. Implementation of these techniques, however, can be challenging for experts and non-experts alike. There is a clear need for software that provides rapid, reliable, and easy access to a wide range of advanced sampling methods and that facilitates implementation of new techniques as they emerge. Here we present SSAGES, a publicly available Software Suite for Advanced General Ensemble Simulations designed to interface with multiple widely used molecular dynamics simulations packages. SSAGES allows facile application of a variety of enhanced sampling techniques—including adaptive biasing force, string methods, and forward flux sampling—that extract meaningful free energy and transition path data from all-atom and coarse-grained simulations. A noteworthy feature of SSAGES is a user-friendly framework that facilitates further development and implementation of new methods and collective variables. In this work, the use of SSAGES is illustrated in the context of simple representative applications involving distinct methods and different collective variables that are available in the current release of the suite. The code may be found at: https://github.com/MICCoM/SSAGES-public.
Water Flux Induced Reorientation of Liquid Crystals
Hadi Ramezani-Dakhel, Monirosadat Sadati, Rui Zhang, Mohammad Rahimi, Khia Kurtenbach, Benoît Roux, and Juan J. de Pablo
ACS Cent. Sci. 3, 1345–1349 (2017). DOI: 10.1021/acscentsci.7b00495
It is well understood that the adsorption of solutes at the interface between a bulk liquid crystal phase and an aqueous phase can lead to orientational or anchoring transitions. A different principle is introduced here, whereby a transient reorientation of a thermotropic liquid crystal is triggered by a spontaneous flux of water across the interface. A critical water flux can be generated by the addition of an electrolyte to the bulk aqueous phase, leading to a change in the solvent activity; water is then transported through the liquid crystal phase and across the interface. The magnitude of the spontaneous water flux can be controlled by the concentration and type of solutes, as well as the rate of salt addition. These results present new, previously unappreciated fundamental principles that could potentially be used for the design of materials involving transient gating mechanisms, including biological sensors, drug delivery systems, separation media, and molecular machines.
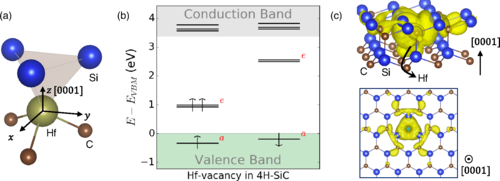
Designing defect-based qubit candidates in wide-gap binary semiconductors for solid-state quantum technologies
Hosung Seo, He Ma, Marco Govoni, and Giulia Galli
Phys. Rev. Materials 1, 075002 (2017). DOI: 0.1103/PhysRevMaterials.1.075002
The development of novel quantum bits is key to extending the scope of solid-state quantum-information science and technology. Using first-principles calculations, we propose that large metal ion–vacancy pairs are promising qubit candidates in two binary crystals: 4H-SiC and w-AlN. In particular, we found that the formation of neutral Hf- and Zr-vacancy pairs is energetically favorable in both solids; these defects have spin-triplet ground states, with electronic structures similar to those of the diamond nitrogen-vacancy center and the SiC divacancy. Interestingly, they exhibit different spin-strain coupling characteristics, and the nature of heavy metal ions may allow for easy defect implantation in desired lattice locations and ensure stability against defect diffusion. To support future experimental identification of the proposed defects, we report predictions of their optical zero-phonon line, zero-field splitting, and hyperfine parameters. The defect design concept identified here may be generalized to other binary semiconductors to facilitate the exploration of new solid-state qubit
Electrostatic confinement and manipulation of DNA molecules for genome analysis
Kristy L. Kounovsky-Shafer, Juan P. Hernandez-Ortiz, Konstantinos Potamousis, Gene Tsvid, Michael Place, Prabu Ravindran, Kyubong Jo, Shiguo Zhou, Theo Odijk, Juan J. de Pablo and David C. Schwartz
PNAS 114, 13400-13405 (2017). DOI: 10.1073/pnas.1711069114
Very large DNA molecules enable comprehensive analysis of complex genomes, such as human, cancer, and plants because they span across sequence repeats and complex somatic events. When physically manipulated, or analyzed as single molecules, long polyelectrolytes are problematic because of mechanical considerations that include shear-mediated breakage, dealing with the massive size of these coils, or the length of stretched DNAs using common experimental techniques and fluidic devices. Accordingly, we harness analyte “issues” as exploitable advantages by our invention and characterization of the “molecular gate,” which controls and synchronizes formation of stretched DNA molecules as DNA dumbbells within nanoslit geometries. Molecular gate geometries comprise micro- and nanoscale features designed to synergize very low ionic strength conditions in ways we show effectively create an “electrostatic bottle.” This effect greatly enhances molecular confinement within large slit geometries and supports facile, synchronized electrokinetic loading of nanoslits, even without dumbbell formation. Device geometries were considered at the molecular and continuum scales through computer simulations, which also guided our efforts to optimize design and functionalities. In addition, we show that the molecular gate may govern DNA separations because DNA molecules can be electrokinetically triggered, by varying applied voltage, to enter slits in a size-dependent manner. Lastly, mapping the Mesoplasma florum genome, via synchronized dumbbell formation, validates our nascent approach as a viable starting point for advanced development that will build an integrated system capable of large-scale genome analysis.
First-principles simulations of heat transport
Marcello Puligheddu, Francois Gygi, and Giulia Galli
Phys. Rev. Materials 1, 060802(R) (2017). DOI: 10.1103/PhysRevMaterials.1.060802
Advances in understanding heat transport in solids were recently reported by both experiment and theory. However an efficient and predictive quantum simulation framework to investigate thermal properties of solids, with the same complexity as classical simulations, has not yet been developed. Here we present a method to compute the thermal conductivity of solids by performing ab initio molecular dynamics at close to equilibrium conditions, which only requires calculations of first-principles trajectories and atomic forces, thus avoiding direct computation of heat currents and energy densities. In addition the method requires much shorter sequential simulation times than ordinary molecular dynamics techniques, making it applicable within density functional theory. We discuss results for a representative oxide, MgO, at different temperatures and for ordered and nanostructured morphologies, showing the performance of the method in different conditions.
Strong Attractions and Repulsions Mediated by Monovalent Salts
Yaohua Li, Martin Girard, Meng Shen, Jaime Andres Millan, and Monica Olvera de la Cruz
PNAS 114, 11838 (2017). DOI: 10.1073/pnas.1713168114
Controlling interactions between proteins and nanoparticles in electrolyte solutions is crucial for advancing biological sciences and biotechnology. The assembly of charged nanoparticles (NPs) and proteins in aqueous solutions can be directed by modifying the salt concentration. High concentrations of monovalent salt can induce the solubilization or crystallization of NPs and proteins. By using a multiscale coarse-grained molecular dynamics approach, we show that, due to ionic correlations in the electrolyte, NPs pairs at high monovalent salt concentrations interact via remarkably strong long-range attractions or repulsions, which can be split into three regimes depending on the surface charge densities of the NPs. NPs with zero-to-low surface charge densities interact via a long-range attraction that is stronger and has a similar range to the depletion attraction induced by polymers with radius of gyrations comparable to the NP diameter. On the other hand, moderately charged NPs with smooth surfaces as well as DNA-functionalized NPs with no possibility of hybridization between them interact via a strong repulsion of range and strength larger than the repulsion predicted by models that neglect ionic correlations, including the Derjaguin–Landau–Vervey–Overbeek (DLVO) model. Interactions between strongly charged NPs (>2 e/nm2), both types smooth and DNA-functionalized NPs, show an attractive potential well at intermediate-to-high salt concentrations, which demonstrates that electrolytes can induce aggregation of strongly charged NPs. Our work provides an improved understanding of the role of ionic correlations in NP assembly and design rules to utilize the salting-out process to crystallize NPs.
In silico evidence for sequence-dependent nucleosome sliding
Joshua Lequieu, David C. Schwartz and Juan J. de Pablo
PNAS 114, E9197 (2017). DOI: 10.1073/pnas.1705685114
Nucleosomes represent the basic building block of chromatin and provide an important mechanism by which cellular processes are controlled. The locations of nucleosomes across the genome are not random but instead depend on both the underlying DNA sequence and the dynamic action of other proteins within the nucleus. These processes are central to cellular function, and the molecular details of the interplay between DNA sequence and nucleosome dynamics remain poorly understood. In this work, we investigate this interplay in detail by relying on a molecular model, which permits development of a comprehensive picture of the underlying free energy surfaces and the corresponding dynamics of nucleosome repositioning. The mechanism of nucleosome repositioning is shown to be strongly linked to DNA sequence and directly related to the binding energy of a given DNA sequence to the histone core. It is also demonstrated that chromatin remodelers can override DNA-sequence preferences by exerting torque, and the histone H4 tail is then identified as a key component by which DNA-sequence, histone modifications, and chromatin remodelers could in fact be coupled.
Surface polarization effects on ion-containing emulsions
Meng Shen, Honghao Li, and Monica Olvera de la Cruz
Phys. Rev. Lett. 119, 138002 (2017). DOI: 10.1103/PhysRevLett.119.138002
Surface polarization in ion-containing heterogeneous dielectric media such as cell media and emulsions is determined by and determines the positions of the ions. We compute the surface polarization self-consistently as the ions move and analyze their effects on the interactions between electro-neutral, ion-containing droplets using coarse-grained molecular dynamics simulations based on the true energy functional. For water droplets immersed in oil, the interdroplet interaction is attractive, and the surface polarization makes the major contribution. By contrast, for oil droplets in water, the ion-surface induced charge interaction is repulsive and counteracts the attraction between the ions, leading to a small attractive interaction between the droplets. This research improves our understanding of self-assembly in mixed phases such as metal extraction for recovering rare earth elements and nuclear waste as well as water purification.
Optimizing surface defects for atomic-scale electronics: Si dangling bonds
Peter Scherpelz and Giulia Galli
Phys. Rev. Materials 1, 021602(R) (2017). DOI: 10.1103/PhysRevMaterials.1.021602
Surface defects created and probed with scanning tunneling microscopes are a promising platform for 1 atomic-scale electronics and quantuminformation technology applications. Using first-principles calculations we demonstrate how to engineer dangling bond (DB) defects on hydrogenated Si(100) surfaces, which give rise to isolated impurity states that can be used in atomic-scale devices. In particular, we show that sample thickness and biaxial strain can serve as control parameters to design the electronic properties of DB defects. While in thick Si samples the neutral DB state is resonant with bulk valence bands, ultrathin samples (1–2 nm) lead to an isolated impurity state in the gap; similar behavior is seen for DB pairs and DB wires. Strain further isolates the DB from the valence band, with the response to strain heavily dependent on sample thickness. These findings suggest new methods for tuning the properties of defects on surfaces for electronic and quantum information applications. Finally, we present a consistent and unifying interpretation of many results presented in the literature for DB defects on hydrogenated silicon surfaces, rationalizing apparent discrepancies between different experiments and simulations.
Parallel O(N) Stokes’ solver towards scalable Brownian dynamics of hydrodynamically interacting objects in general geometries
Xujun Zhao, Jiyuan Li, Xikai Jiang, Dmitry Karpeev, Olle Heinonen, Barry Smith, Juan P. Hernandez-Ortiz, and Juan J. de Pablo
J. Chem. Phys. 146, 244114 (2017). DOI: 10.1063/1.4989545
An efficient parallel Stokes’s solver has been developed for complete description of hydrodynamic interactions between Brownian particles in bulk and confined geomteries. A Langevin description of the particle dynamics is adopted, where the long-range interactions are included using a Green’s function formalism. A scalable parallel computational approach is presented, where the general geometry Stokeslet is calculated following a matrix-free highly-efficient iterative finite-element Stokes’ solver for the accurate treatment of long-range hydrodynamic interactions in arbitrary confined geometries. A combination of mid-point time integration of the Brownian stochastic differential equation, the parallel Stokes’ solver, and a Chebyshev polynomial approximation for the fluctuation-dissipation theorem lead to an O(N) parallel algorithm. We illustrate the new algorithm in the context of the dynamics of confined polymer solutions under equilibrium and non-equilibrium conditions. The method is then extended to treat suspended finite size particles of arbitrary shape in any geometry using an Immersed Boundary approach.

The Emergent Nematic Phase of Ionic Chromonic Liquid Crystals
Hythem Sidky and Jonathan K. Whitmer
J. Phys. Chem. B. 121, 6691-6698 (2017). DOI: 10.1021/acs.jpcb.7b03113
Chromonic liquid crystals exhibit a unique self-assembly process which is of both theoretical and practical interest. A characteristic feature of chromonics is the occurrence of molecular association through stacking at extremely low concentrations. Experimental evidence has suggested that this process is approximately isodesmic across a broad concentration range. Another important aspect of these phases is a separation of energy scales for elastic deformation that leads to novel bulk morphologies and defect arrangements. To date, only a handful of computational studies have managed to reproduce crucial aspects of chromonic phases, with ionic chromonics treated only by expensive fully atomistic simulations. Here, we present a simple model based on the competition between long-range repulsions and short-range anisotropic attractions capable of capturing all features of the chromonic phase. Molecular simulations of coarse-grained mesogens are used to map out the phase behavior and explore how structural and energetic anisotropies influence their ordering and response. This work presents the first computational investigation into the nematic elasticity of these phases and demonstrates key correlations between elastic response and stack growth.

Electronic Structure of Aqueous Solutions: Bridging the Gap Between Theory and Experiments
Tuan Anh Pham, Marco Govoni, Robert Seidel, Stephen E. Bradforth, Eric Schwegler, and Giulia Galli
Sci. Adv. 3, 1603210 (2017). DOI: 10.1126/sciadv.1603210
Predicting the electronic properties of aqueous liquids has been a long-standing challenge for quantum mechanical methods. Yet it is a crucial step in understanding and predicting the key role played by aqueous solutions and electrolytes in a wide variety of emerging energy and environmental technologies, including battery and photoelectrochemical cell design. Here we propose an efficient and accurate approach to predict the electronic properties of aqueous solutions, based on the combination of first-principles methods and experimental validation using state-of-the-art spectroscopic measurements. We present results of the photoelectron spectra of a broad range of solvated ions, showing that first-principles molecular dynamics simulations and electronic structure calculations using dielectric hybrid functionals provide a quantitative description of the electronic properties of the solvent and solutes, including excitation energies. The proposed computational framework is general and applicable to other liquids, thereby offering great promise in understanding and engineering solutions and liquid electrolytes for a variety of important energy technologies.

Performance and self-consistency of the generalized dielectric dependent hybrid functional
Nicholas P. Brawand, Marco Govoni, Márton Vörös, and Giulia Galli
J. Chem. Theory Comput. 13, 3318 (2017), DOI: 10.1021/acs.jctc.7b00368
We analyze the performance of the recently proposed screened exchange constant functional (SX) (Phys. Rev. X 2016, 10, 642) on the GW100 test set, and we discuss results obtained at different levels of self-consistency. The SX functional is a generalization of dielectric dependent hybrid functionals to finite systems, it is non-empirical and depends on the average screening of the exchange interaction. We compare results for ionization potentials obtained with SX to those of CCSD(T) calculations and experiments and we find excellent agreement, on par with recent state of the art methods based on many body perturbation theory. Applying SX perturbatively to correct PBE eigenvalues yields improved results in most cases, except for ionic molecules, for which wave function self-consistency is instead crucial. Calculations where wave functions and the screened exchange constant (αSX) are determined self-consistently, and those where αSX is fixed to the value determined within PBE, yield results of comparable accuracy. Perturbative G0W0 corrections of eigenvalues obtained with self-consistent αSX are small on average, for all molecules in the GW100 test set.
Charge Transport in Nanostructured Materials: Implementation and Verification of Constrained Density Functional Theory
Matthew Goldey, Nicholas Brawand, Márton Vörös, and Giulia Galli
J. Chem. Theory Comput., 13, 2581-2590 (2017). DOI: 10.1021/acs.jctc.7b00088
The in silico design of novel complex materials for energy conversion requires accurate, ab initio simulation of charge transport. In this work, we present an implementation of constrained density functional theory (CDFT) for the calculation of parameters for charge transport in the hopping regime. We verify our implementation against literature results for molecular systems, and we discuss the dependence of results on numerical parameters and the choice of localization potentials. In addition, we compare CDFT results with those of other commonly used methods for simulating charge transport between nanoscale building blocks. We show that some of these methods give unphysical results for thermally disordered configurations, while CDFT proves to be a viable and robust approach.
Influence of Vapor Deposition on Structural and Charge Transport Properties of Ethylbenzene Films
Lucas W. Antony, Nicholas E. Jackson, Ivan Lyubimov, Venkatram Vishwanath, Mark D. Ediger, and Juan J. de Pablo
ACS Central Science, 3, 415-424 (2017). DOI: 10.1021/acscentsci.7b00041
Organic glass films formed by physical vapor deposition exhibit enhanced stability relative to those formed by conventional liquid cooling and aging techniques. Recently, experimental and computational evidence has emerged indicating that the average molecular orientation can be tuned by controlling the substrate temperature at which these “stable glasses” are grown. In this work, we present a comprehensive all-atom simulation study of ethylbenzene, a canonical stable-glass former, using a computational film formation procedure that closely mimics the vapor deposition process. Atomistic studies of experimentally formed vapor-deposited glasses have not been performed before, and this study therefore begins by verifying that the model and method utilized here reproduces key structural features observed experimentally. Having established agreement between several simulated and experimental macroscopic observables, simulations are used to examine the substrate temperature dependence of molecular orientation. The results indicate that ethylbenzene glasses are anisotropic, depending upon substrate temperature, and that this dependence can be understood from the orientation present at the surface of the equilibrium liquid. By treating ethylbenzene as a simple model for molecular semiconducting materials, a quantum-chemical analysis is then used to show that the vapor-deposited glasses exhibit decreased energetic disorder and increased magnitude of the mean-squared transfer integral relative to isotropic, liquid-cooled films, an effect that is attributed to the anisotropic ordering of the molecular film. These results suggest a novel structure–function simulation strategy capable of tuning the electronic properties of organic semiconducting glasses prior to experimental deposition, which could have considerable potential for organic electronic materials design.

Design of heterogeneous chalcogenide nanostructures with pressure-tunable gaps and without electronic trap states
Federico Giberti, Márton Vörös, and Giulia Galli
Nano Lett. 17, 2547–2553 (2017). DOI: 10.1021/acs.nanolett.7b00283
Heterogeneous nanostructures, such as quantum dots (QDs) embedded in solid matrices or core–shell nanoparticles, are promising platforms for a wide variety of applications, including phosphors with increased quantum yield, photocatalysis, and solar energy conversion. However, characterizing and controlling their interfacial morphology and defects, which greatly influence their electronic properties, have proven difficult in numerous cases. Here we carried out atomistic calculations on chalcogenide nanostructured materials, i.e., PbSe QDs in CdSe matrices and CdSe embedded in PbSe, and we established how interfacial and core structures affect their electronic properties. In particular, we showed that defects present at interfaces of PbSe nanoparticles and CdSe matrices give rise to detrimental intragap states, degrading the performance of photovoltaic devices. Instead, the electronic gaps of the inverted system (CdSe dots in PbSe) are clean, indicating that this material has superior electronic properties for solar applications. In addition, our calculations predicted that the core structure of CdSe and in turn its band gap may be tuned by applying pressure to the PbSe matrix, providing a means to engineering the properties of new functional materials.
Understanding Atomic-Scale Behavior of Liquid Crystals at Aqueous Interfaces
Hadi Ramezani-Dakhel, Monirosadat Sadati, Mohammad Rahimi, Abelardo Ramirez-Hernandez, Benoît Roux, and Juan J. de Pablo
J. Chem. Theory Comput. 13, 237–244 (2017). DOI: 10.1021/acs.jctc.6b00844
The ordered environment presented by liquid crystals at interfaces enables a range of novel functionalities that is only now beginning to be exploited in applications ranging from light focusing devices to biosensors. One key feature of liquid crystals is that molecular events occurring at an interface propagate over large distances through the bulk. In spite of their importance, our fundamental understanding of liquid crystal–water and liquid crystal–air interfaces remains limited. In this work, we present results from large-scale atomistic molecular dynamics simulations on the organization of the nematic and isotropic phases of the nitrile-containing mesogenic molecule 4-cyano-4′-pentylbiphenyl (5CB) in the vicinity of vacuum and aqueous interfaces. Hybrid boundary conditions are imposed by confining 5CB films between vacuum and an aqueous medium to examine how those two types of interfaces influence the specific structural arrangement and ordering of 5CB. Consistent with experiments, our results indicate that 5CB exhibits homeotropic anchoring at the vacuum interface, and planar alignment at aqueous interfaces. Two-dimensional molecular dynamics potential of mean force calculations and average polarization densities show that the polar nitrile group of 5CB remains hydrated near the aqueous interface, where it modulates the orientation of water molecules. Estimates of the anchoring strength reveal an oscillatory decay and a semilinear decay with distance from the interface in vacuum and water, respectively.
Molecular Structure of Canonical Liquid Crystal Interfaces
Monirosadat Sadati, Hadi Ramezani-Dakhel, Wei Bu, Emre Sevgen, Zhu Liang, Cem Erol, Mohammad Rahimi, Nader Taheri Qazvini, Binhua Lin, Nicholas L. Abbott, Benoı̂t Roux, Mark L. Schlossman, and Juan J. de Pablo
J. Am. Chem. Soc. 139, 3841–3850 (2017). DOI: 10.1021/jacs.7b00167
Numerous applications of liquid crystals rely on control of molecular orientation at an interface. However, little is known about the precise molecular structure of such interfaces. In this work, synchrotron X-ray reflectivity measurements, accompanied by large-scale atomistic molecular dynamics simulations, are used for the first time to reconstruct the air-liquid crystal interface of a nematic material, namely, 4-pentyl-4′-cyanobiphenyl (5CB). The results are compared to those for 4-octyl-4′-cyanobiphenyl (8CB) which, in addition to adopting isotropic and nematic states, can also form a smectic phase. Our findings indicate that the air interface imprints a highly ordered structure into the material; such a local structure then propagates well into the bulk of the liquid crystal, particularly for nematic and smectic phases.

Local and Global Effects of Dissolved Sodium Chloride on the Structure of Water
Alex P. Gaiduk and Giulia Galli
J. Phys. Chem. Lett. 8, 1496–1502 (2017). DOI: 10.1021/acs.jpclett.7b00239
Determining how the structure of water is modified by the presence of salts is instrumental to understanding the solvation of biomolecules and, in general, the role played by salts in biochemical processes. However, the extent of hydrogen bonding disruption induced by salts remains controversial. We performed extensive first-principles simulations of solutions of a simple salt (NaCl) and found that, while the cation does not significantly change the structure of water beyond the first solvation shell, the anion has a further reaching effect, modifying the hydrogen-bond network even outside its second solvation shell. We found that a distinctive fingerprint of hydrogen bonding modification is the change in polarizability of water molecules. Molecular dipole moments are instead insensitive probes of long-range modifications induced by Na+ and Cl– ions. Though noticeable, the long-range effect of Cl– is expected to be too weak to affect solubility of large biomolecules.
New Forms of CdSe: Molecular Wires, Gels, and Ordered Mesoporous Assemblies
Margaret Hudson, Dmitriy Dolzhnikov, Alexander S. Filatov, Eric M. Janke, Jaeyoung Jang, Byeongdu Lee, Chengjun Sun, and Dmitri Talapin
J. Am. Chem. Soc. 139, 3368-3377 (2017). DOI: 10.1021/jacs.6b10077
This work investigates the structure and properties of soluble chalcogenidocadmates, a molecular form of cadmium chalcogenides with unprecedented one-dimensional bonding motifs. The single crystal X-ray structure reveals that sodium selenocadmate consists of infinite one-dimensional wires of (Cd2Se3)n2n– charge balanced by Na+ and stabilized by coordinating solvent molecules. Exchanging the sodium cation with tetraethylammonium or didodecyldimethylammonium expands the versatility of selenocadmate by improving its solubility in a variety of polar and nonpolar solvents without changing the anion structure and properties. The introduction of a micelle-forming cationic surfactant allows for the templating of selenocadmate, or the analogous telluride species, to create ordered organic–inorganic hybrid CdSe or CdTe mesostructures. Finally, the interaction of selenocadmate “wires” with Cd2+ ions creates an unprecedented gel-like form of stoichiometric CdSe. We also demonstrate that these low-dimensional CdSe species show characteristic semiconductor behavior, and can be used in photodetectors and field-effect transistors.
Instability and Efficiency of Mixed Halide Perovskites CH3NH3AI3-xClx (A=Pb and Sn): a first principles, computational study
Yuping He and Giulia Galli
Chem. Mater. 29, 682–689 (2017). DOI: 10.1021/acs.chemmater.6b04300
We carried out calculations based on density functional theory to investigate the electronic, vibrational, and dielectric properties of mixed halide perovskites CH3NH3 AI3–xClx with A = Pb and Sn. Computed free energies indicated that Cl mixed systems may be formed only for Cl concentrations not exceeding 1019 cm–3, and phonon calculations showed that the disorder induced in the host lattice by the presence of a smaller halogen is responsible for mechanical instabilities. However, we found that the presence of chloride may be beneficial to the electronic properties of the perovskites. Chloride anions cause the organic cations to be displaced from the center of the cage; such a displacement induces preferential orientations of the cation dipole, which in turn are responsible for notable changes in the dielectric properties of the material and possibly for the formation of local ferroelectric domains. The latter are instrumental in separating electron hole pairs and hence in contributing to long charge-carrier diffusion lengths, in spite of polarons being more likely formed in mixed perovksites than in CH3NH3 AI3.
Age and Structure of a Model Vapor-Deposited Glass
Daniel Reid, Ivan Lyubimov, M. D. Ediger, and Juan J. de Pablo
Nat. Commun. 7, 13062 (2016). DOI: 10.1038/ncomms13062
Glass films prepared by a process of physical vapour deposition have been shown to have thermodynamic and kinetic stability comparable to those of ordinary glasses aged for thousands of years. A central question in the study of vapour-deposited glasses, particularly in light of new knowledge regarding anisotropy in these materials, is whether the ultra-stable glassy films formed by vapour deposition are ever equivalent to those obtained by liquid cooling. Here we present a computational study of vapour deposition for a two-dimensional glass forming liquid using a methodology, which closely mimics experiment. We find that for the model considered here, structures that arise in vapour-deposited materials are statistically identical to those observed in ordinary glasses, provided the two are compared at the same inherent structure energy. We also find that newly deposited hot molecules produce cascades of hot particles that propagate far into the film, possibly influencing the relaxation of the material.
Generalization of Dielectric-Dependent Hybrid Functionals to Finite Systems
Nicholas P. Brawand, Márton Vörös, Marco Govoni, and Giulia Galli
Phys. Rev. X 6, 041002 (2016). DOI: 10.1103/PhysRevX.6.041002
The accurate prediction of electronic and optical properties of molecules and solids is a persistent challenge for methods based on density functional theory. We propose a generalization of dielectric-dependent hybrid functionals to finite systems where the definition of the mixing fraction of exact and semilocal exchange is physically motivated, nonempirical, and system dependent. The proposed functional yields ionization potentials, and fundamental and optical gaps of many, diverse molecular systems in excellent agreement with experiments, including organic and inorganic molecules and semiconducting nanocrystals. We further demonstrate that this hybrid functional gives the correct alignment between energy levels of the exemplary TTF-TCNQ donor-acceptor system.

Self-Assembly of Colloidal Nanocrystals: From Intricate Structures to Functional Materials
Michael A. Boles, Michael Engel, and Dmitri V. Talapin
Chem. Rev. 116, 11220–11289 (2016). DOI: 0.1021/acs.chemrev.6b00196
Chemical methods developed over the past two decades enable preparation of colloidal nanocrystals with uniform size and shape. These Brownian objects readily order into superlattices. Recently, the range of accessible inorganic cores and tunable surface chemistries dramatically increased, expanding the set of nanocrystal arrangements experimentally attainable. In this review, we discuss efforts to create next-generation materials via bottom-up organization of nanocrystals with preprogrammed functionality and self-assembly instructions. This process is often driven by both interparticle interactions and the influence of the assembly environment. The introduction provides the reader with a practical overview of nanocrystal synthesis, self-assembly, and superlattice characterization. We then summarize the theory of nanocrystal interactions and examine fundamental principles governing nanocrystal self-assembly from hard and soft particle perspectives borrowed from the comparatively established fields of micrometer colloids and block copolymer assembly. We outline the extensive catalog of superlattices prepared to date using hydrocarbon-capped nanocrystals with spherical, polyhedral, rod, plate, and branched inorganic core shapes, as well as those obtained by mixing combinations thereof. We also provide an overview of structural defects in nanocrystal superlattices. We then explore the unique possibilities offered by leveraging nontraditional surface chemistries and assembly environments to control superlattice structure and produce nonbulk assemblies. We end with a discussion of the unique optical, magnetic, electronic, and catalytic properties of ordered nanocrystal superlattices, and the coming advances required to make use of this new class of solids.
Intermolecular Structural Change for Thermoswitchable Polymeric Photosensitizer
Wooram Park, Sin-Jung Park, Soojeong Cho, Heejun Shin, Young-Seok Jung, Byeongdu Lee, Kun Na, and Dong-Hyun Kim
J. Am. Chem. Soc. 138, 10734-10737 (2016). DOI: 10.1021/jacs.6b04875
We developed a thermoswitchable polymeric photosensitizer (T-PPS) by conjugating PS (Pheophorbide-a, PPb-a) to a temperature-responsive polymer backbone of biocompatible hydroxypropyl cellulose. Self-quenched PS molecules linked in close proximity by π–π stacking in T-PPS were easily transited to an active monomeric state by the temperature-induced phase transition of polymer backbones. The temperature-responsive intermolecular interaction changes of PS molecules in T-PPS were demonstrated in synchrotron small-angle X-ray scattering and UV–vis spectrophotometer analysis. The T-PPS allowed switchable activation and synergistically enhanced cancer cell killing effect at the hyperthermia temperature (45 °C). Our developed T-PPS has the considerable potential not only as a new class of photomedicine in clinics but also as a biosensor based on temperature responsiveness.
Mechanical Response of DNA–Nanoparticle Crystals to Controlled Deformation
Joshua Lequieu, Andrés Córdoba, Daniel Hinckley, and Juan J. de Pablo
ACS Cent. Sci. 2, 614-620 (2016). DOI: 10.1021/acscentsci.6b00170
The self-assembly of DNA-conjugated nanoparticles represents a promising avenue toward the design of engineered hierarchical materials. By using DNA to encode nanoscale interactions, macroscale crystals can be formed with mechanical properties that can, at least in principle, be tuned. Here we present in silico evidence that the mechanical response of these assemblies can indeed be controlled, and that subtle modifications of the linking DNA sequences can change the Young’s modulus from 97 kPa to 2.1 MPa. We rely on a detailed molecular model to quantify the energetics of DNA–nanoparticle assembly and demonstrate that the mechanical response is governed by entropic, rather than enthalpic, contributions and that the response of the entire network can be estimated from the elastic properties of an individual nanoparticle. The results here provide a first step toward the mechanical characterization of DNA–nanoparticle assemblies, and suggest the possibility of mechanical metamaterials constructed using DNA.
Tension-Dependent Free Energies of Nucleosome Unwrapping
Joshua Lequieu, Andrés Córdoba, David C. Schwartz, and Juan J. de Pablo
ACS Cent. Sci. 2, 660-666 (2016). DOI: 10.1021/acscentsci.6b00201
Nucleosomes form the basic unit of compaction within eukaryotic genomes, and their locations represent an important, yet poorly understood, mechanism of genetic regulation. Quantifying the strength of interactions within the nucleosome is a central problem in biophysics and is critical to understanding how nucleosome positions influence gene expression. By comparing to single-molecule experiments, we demonstrate that a coarse-grained molecular model of the nucleosome can reproduce key aspects of nucleosome unwrapping. Using detailed simulations of DNA and histone proteins, we calculate the tension-dependent free energy surface corresponding to the unwrapping process. The model reproduces quantitatively the forces required to unwrap the nucleosome and reveals the role played by electrostatic interactions during this process. We then demonstrate that histone modifications and DNA sequence can have significant effects on the energies of nucleosome formation. Most notably, we show that histone tails contribute asymmetrically to the stability of the outer and inner turn of nucleosomal DNA and that depending on which histone tails are modified, the tension-dependent response is modulated differently.
An O(N) and parallel approach to integral problems by a kernel-independent fast multipole method: Application to polarization and magnetization of interacting particles
Xikai Jiang, Jiyuan Li, Xujun Zhao, Jian Qin, Dmitry Karpeev, Juan Hernandez-Ortiz, Juan J. de Pablo,and Olle Heinonen
J. Chem. Phys. 145, 064307 (2016). DOI: 10.1063/1.4960436
Large classes of materials systems in physics and engineering are governed by magnetic and electrostatic interactions. Continuum or mesoscale descriptions of such systems can be cast in terms of integral equations, whose direct computational evaluation requires O(N2) operations, where N is the number of unknowns. Such a scaling, which arises from the many-body nature of the relevant Green’s function, has precluded wide-spread adoption of integral methods for solution of large-scale scientific and engineering problems. In this work, a parallel computational approach is presented that relies on using scalable open source libraries and utilizes a kernel-independent Fast Multipole Method (FMM) to evaluate the integrals in O(N) operations, with O(N) memory cost, thereby substantially improving the scalability and efficiency of computational integral methods. We demonstrate the accuracy, efficiency, and scalability of our approach in the context of two examples. In the first, we solve a boundary value problem for a ferroelectric/ferromagnetic volume in free space. In the second, we solve an electrostatic problem involving polarizable dielectric bodies in an unbounded dielectric medium. The results from these test cases show that our proposed parallel approach, which is built on a kernel-independent FMM, can enable highly efficient and accurate simulations and allow for considerable flexibility in a broad range of applications.
A coarse-grained thermodynamic model for the predictive engineering of valence-selective membranes
Vikramjit Singh Rathee, Siyi Qu, William A. Phillip, and Jonathan K. Whitmer
Mol. Syst. Des. Eng. 1, 301-312 (2016). DOI: 10.1039/C6ME00045B
Membrane-based filtration has shown promise in the treatment of municipal and industrial water supplies. Most existing filtration membranes rely on size-selective rejection mechanisms and do not offer a systematic means to tune the chemistry of the membrane and, in turn, the rejection of solutes based on chemical factors. Technological advancements in copolymer self-assembly have permitted the creation of highly porous membranes through arrested microphase separation. The resulting structures exhibit nanoscale pores whose size and chemistry are controlled by the macromolecular architecture of the copolymer precursors. Here, we examine in detail how the structure and chemistry of charge-functionalized copolymers affect their performance as nanofiltration membranes through experiments and coarse-grained molecular simulation of membranes formed from poly(acrylonitrile-r-oligo(ethylene glycol) methyl ether methacrylate-r-glycidyl methacrylate) (P(AN-r-OEGMA-r-GMA)). Charge-selective moieties were introduced into the membranes through reactions with diamine salts of varying length, enabling these membranes to offer preferential rejection of multivalent ionic species. Using a minimal model, we explore the essential thermodynamics features of salt rejection in these membranes, and develop a model capable of linking the performance of the membranes to their molecular character through measurement of the underlying free energy profile. Our key result is an effective membrane porosity which quantitatively describes experiments for a wide variety of situations after calibrating a single parameter. These results demonstrate that at moderate ion concentrations and flow rates, the selection of ions is driven predominantly by equilibrium thermodynamics. Going forward, the ability of the model to capture the rejection of dissolved solutes can be leveraged to design optimized membranes for targeted performance profiles in silico before undertaking time consuming batteries of Edisonian experiments.
Elastic properties of common Gay–Berne nematogens from density of states (DOS) simulations
Hythem Sidky and Jonathan K. Whitmer
Liquid Crystals 43, 2285-2299 (2016). DOI: 10.1080/02678292.2016.1201869
A free energy perturbation method is used to systematically study the elastic properties of four common Gay–Berne nematogenic models; two with a length-to-diameter ratio κ = 3 [(3, 5, 1, 2) and (3, 5, 1, 3)], a model with κ = 4.4 parameterised for p-terphenyl (4.4, 20, 1, 1), and a discogen with κ = 0.345 (0.345, 0.2, 1, 2). In accordance with previous measurements, we find that for κ = 3, models, . We additionally find the latter two models in particular accurately capture the experimentally measured elastic ratios in apolar achiral systems. The (4.4, 20, 1, 1) model reproduces the elastic constant ratios of p-azoxyanisole remarkably well, and maps to within 30% of the absolute. The (0.345, 0.2, 1, 2) model elastic constants exhibit an unusual temperature dependence similar to recent experimental studies. Here we find , in line with theoretical predictions. All models deviate from the mean-field expectation kii ∝ S2. These results represent a crucial first step towards quantitatively accurate coarse-grained liquid crystalline models of self-assembly and response, enabling one to choose a Gay–Berne model based on its measured elastic ratios rather than just its shape and energy anisotropy.
Implementation and Validation of Fully-Relativistic GW Calculations: Spin-Orbit Coupling in Molecules, Nanocrystals and Solids
Peter Scherpelz, Marco Govoni, Ikutaro Hamada, and Giulia Galli
J. Chem. Theory Comput. 12, 3523-3544 (2016). DOI: 10.1021/acs.jctc.6b00114
We present an implementation of G0W0 calculations including spin–orbit coupling (SOC) enabling investigations of large systems, with thousands of electrons, and we discuss results for molecules, solids, and nanocrystals. Using a newly developed set of molecules with heavy elements (called GW-SOC81), we find that, when based upon hybrid density functional calculations, fully relativistic (FR) and scalar-relativistic (SR) G0W0 calculations of vertical ionization potentials both yield excellent performance compared to experiment, with errors below 1.9%. We demonstrate that while SR calculations have higher random errors, FR calculations systematically underestimate the VIP by 0.1 to 0.2 eV. We further verify that SOC effects may be well approximated at the FR density functional level and then added to SR G0W0 results for a broad class of systems. We also address the use of different root-finding algorithms for the G0W0 quasiparticle equation and the significant influence of including d electrons in the valence partition of the pseudopotential for G0W0 calculations. Finally, we present statistical analyses of our data, highlighting the importance of separating definitive improvements from those that may occur by chance due to a limited number of samples. We suggest the statistical analyses used here will be useful in the assessment of the accuracy of a large variety of electronic structure methods.
Elastic response and phase behavior in binary liquid crystal mixtures
Hythem Sidky and Jonathan K. Whitmer
Soft Matter 12, 4489-4498 (2016). DOI: 10.1039/C5SM03107A
Utilizing density-of-states simulations, we perform a full mapping of the phase behavior and elastic responses of binary liquid crystalline mixtures represented by the multicomponent Lebwohl–Lasher model. Our techniques are able to characterize the complete phase diagram, including nematic–nematic phase separation predicted by mean-field theories, but previously not observed in simulations. Mapping this phase diagram permits detailed study of elastic properties across the miscible nematic region. Importantly, we observe for the first time local phase separation and disordering driven by the application of small linear perturbations near the transition temperature and more significantly through nonlinear stresses. These findings are of key importance in systems of blended nematics which contain particulate inclusions, or are otherwise confined.
Photoelectron spectra of aqueous solutions from first principles
Alex P. Gaiduk, Marco Govoni, Robert Seidel, Jonathan Skone, Bernd Winter, and Giulia Galli
J. Am. Chem. Soc. 138, 6912-6915 (2016). DOI: 10.1021/jacs.6b00225
We present a combined computational and experimental study of the photoelectron spectrum of a simple aqueous solution of NaCl. Measurements were conducted on microjets, and first-principles calculations were performed using hybrid functionals and many-body perturbation theory at the G0W0 level, starting with wave functions computed in ab initio molecular dynamics simulations. We show excellent agreement between theory and experiments for the positions of both the solute and solvent excitation energies on an absolute energy scale and for peak intensities. The best comparison was obtained using wave functions obtained with dielectric-dependent self-consistent and range-separated hybrid functionals. Our computational protocol opens the way to accurate, predictive calculations of the electronic properties of electrolytes, of interest to a variety of energy problems.
Lattice Boltzmann simulation of asymmetric flow in nematic liquid crystals with finite anchoring
Rui Zhang, Tyler Roberts, Igor S. Aranson, and Juan J. de Pablo
J. Chem. Phys. 144, 084905 (2016). DOI: 10.1063/1.4940342
Liquid crystals (LCs) display many of the flow characteristics of liquids but exhibit long range orientational order. In the nematic phase, the coupling of structure and flow leads to complex hydrodynamic effects that remain to be fully elucidated. Here, we consider the hydrodynamics of a nematic LC in a hybrid cell, where opposite walls have conflicting anchoring boundary conditions, and we employ a 3D lattice Boltzmann method to simulate the time-dependent flow patterns that can arise. Due to the symmetry breaking of the director field within the hybrid cell, we observe that at low to moderate shear rates, the volumetric flow rate under Couette and Poiseuille flows is different for opposite flow directions. At high shear rates, the director field may undergo a topological transition which leads to symmetric flows. By applying an oscillatory pressure gradient to the channel, a net volumetric flow rate is found to depend on the magnitude and frequency of the oscillation, as well as the anchoring strength. Taken together, our findings suggest several intriguing new applications for LCs in microfluidic devices.